Fig. 1.1
Schematic illustration of the general principles of the stepwise development of a local vascular circuit within extensive mesenchymal spaces. The main steps are (1) formation of a primary vascular plexus, (2) plexus remodeling, and (3) maturation of the vessel wall
During subsequent development, the originally isolated BV-islands fuse with neighboring BV-islands. Consequently, a primitive vascular plexus is formed, which is frequently named the “capillary vascular plexus” since the wall of its vascular meshes consists only of endothelial cells. We should note, however, that the vessel diameters found in primitive embryonic vascular plexuses are larger than the diameters of most of the mature capillaries. Therefore, if we compare such plexuses with vascular beds of the mature human body, we will find that they have more resemblance with the sinusoidal vessels of the adult spleen or bone marrow than with typical capillary beds. We, therefore, think that the term capillary vascular plexus is a misnomer that may provide a wrong picture of important components of the embryonic vasculature. We prefer usage of the term “primary vascular plexus.”
The extent and configuration of a given primary vascular plexus seem to correspond to the spatial configuration of the local mesenchymal space. In cases of extensive mesenchymal spaces, such as the yolk sac mesenchyme, we usually find extensive vascular networks that do not show main vascular channels before the onset of blood flow (Fig. 1.1). In cases of narrow mesenchymal spaces, such as the pharyngeal arches, we usually find narrow vascular plexuses (Fig. 1.2). The anatomy of such plexuses is best described as a “primary plexiform vascular channel” rather than a vascular network. Primary plexiform vascular channels represent the direct forerunners of some of the main arterial or venous trunks of the embryo (e.g., pharyngeal arch arteries, cranial portions of the paired dorsal aortae), while primary vascular plexuses become remodeled into hierarchically arranged local vascular circuits consisting of arteries, veins, and capillaries.
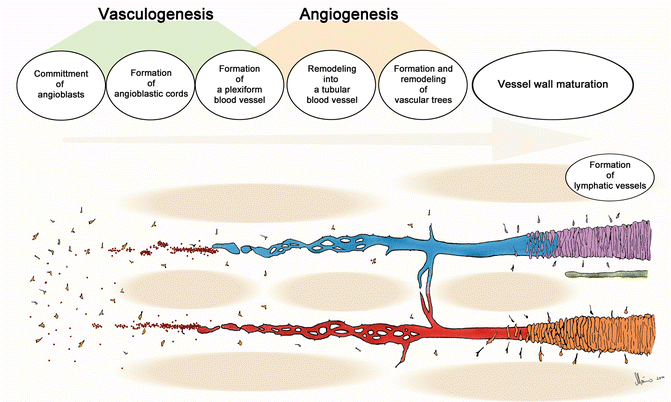
Fig. 1.2
Schematic illustration of the general principles of the formation of arterial and venous channels within narrow mesenchymal spaces. The main steps are (1) formation of primary plexiform vascular channels, (2) channel remodeling, and (3) maturation of the vessel wall
In the initial state, primary vascular plexuses are not perfused with blood, and visual examination does not facilitate the identification of the future course of arterial or venous trunks within the meshes of the vascular network. However, molecular biological analyses have shown that the endothelial cells of the plexuses are already specified for arterial and venous fates. However, the factors causing the arterial-venous pre-patterning of primary vascular plexuses are unknown at the present time [20, 21].
Remodeling of Primary Vascular Plexuses into Hierarchically Arranged Vascular Circuits
As already noted, primary vascular plexuses lack externally visible signs for a hierarchical arrangement of their vascular meshes. The situation changes, however, when a primary vascular plexus becomes connected to already perfused blood vessels. Subsequent to the establishment of permanent blood flow through a primary vascular plexus, the plexus undergoes a process of vascular remodeling, which leads to the establishment of a hierarchically arranged vascular bed consisting of large vascular channels, representing arteries and veins, and of small vascular channels representing capillaries. The wall of all these vessels still consists only of endothelial cell, but their arterial, venous, or capillary identities can be deduced from their topography and the direction of blood flow. During remodeling, the arterial and venous channels first appear as plexiform vascular channels before they assume the phenotype of tubular vessels (Figs. 1.1 and 1.3).
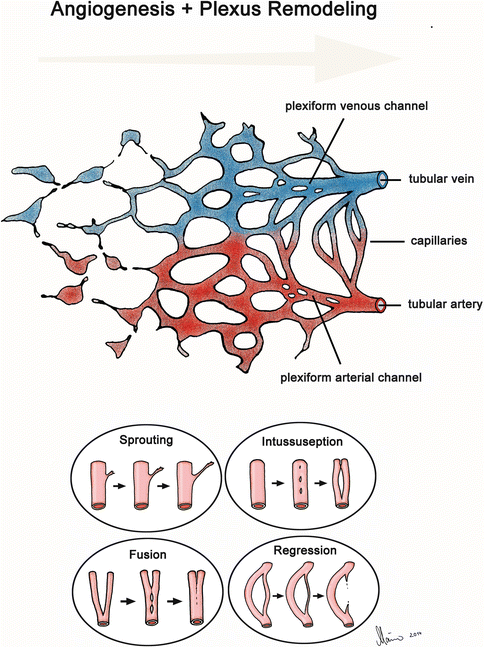
Fig. 1.3
Schematic illustration of the development of a hierarchical vascular tree. (1) Remodeling of the primary vascular plexus into a hierarchically arranged vascular circuit consisting of arteries, capillaries, and veins. (2) Four distinct morphogenetic mechanisms contributing to the remodeling process
Experimental studies on avian embryos (chick, quail) and zebrafish mutants have shown that the prevention of blood flow through primary vascular plexuses prevent the remodeling of such plexuses into hierarchically arranged vascular systems [22–24]. Moreover, experimentally induced changes in the normal blood flow changed the initial arterial-venous pre-patterning of the plexuses [23, 25]. These data suggest that hemodynamic factors play fundamental roles in the remodeling of primary vascular plexuses as well as in the specification of arteries and veins [24, 25].
Remodeling of primary vascular plexuses is accomplished by a set of various morphogenetic mechanisms, which facilitate locally confined changes in size and number of the vascular meshes (Fig. 1.3). The number of originally existing vascular meshes is reduced either by fusion of two neighboring vessels or by the regression of vessels. Vasculogenesis may still contribute to an increase in the number of vascular meshes of the plexus. However, the majority of new blood vessels that become added to a plexus now are generated either by the formation of vascular sprouts that grow out from already existing vessels or from the splitting of already existing vessels. The formation of new blood vessels from preexisting vessels is generally termed “angiogenesis” [12, 13]. Consequently, the abovementioned mechanisms have been named “sprouting angiogenesis,” “splitting angiogenesis,” and “intussusceptive angiogenesis.”
Vessel Wall Maturation
The formation of primary vascular plexuses (Fig. 1.1), primary plexiform vascular channels (Fig. 1.2), as well as plexus remodeling (Fig. 1.3) is induced and controlled by an interplay of genetic factors, such as the expression of regulatory genes and signaling molecules [21, 26–28], and epigenetic factors, such as oxygen [29], metabolites [30], and hemodynamic loads [24, 25, 31]. During embryonic development, all these factors continuously change in a highly dynamic fashion. Corresponding to the continuously changing genetic and epigenetic landscape of the embryo, the developing cardiovascular system undergoes continuous growth and remodeling, which is facilitated by the primary endothelial structure of its vessel walls. At a certain size, however, purely endothelial vessel walls cannot withstand the continuously increasing hemodynamic loads (e.g., blood pressure) acting on the walls of main vascular channels. At this stage of blood vessel development, the walls of the main arterial and venous channels become reinforced by the recruitment of vascular smooth muscle cells (VSMCs) and fibroblasts as well as by the synthesis of extracellular matrix components such as collagen and elastin [32]. This leads to the establishment of the well-known, multilayered wall structure of mature arteries and veins. The establishment of the mature arterial wall structure generally proceeds in a proximal-to-distal direction suggesting that hemodynamic factors play fundamental roles in the maturation of blood vessels [24, 31, 32].
Fate mapping studies on avian (chick, quail) and mammalian (mouse) embryos have shown that the VSMCs forming the tunica media of the aorta and its main branches derive from various embryonic sources (Fig. 1.4) [33, 34]: (1) The so-called secondary heart field provides VSMCs that colonize the roots of the aorta and pulmonary trunk [35]. (2) The “cephalic neural crest” provides VSMCs forming the tunica media of the pulmonary trunk and ductus arteriosus, the ascending aorta and aortic arch, and the brachiocephalic trunk and common carotid arteries [36–39]. (3) The ventral portions of the “somites” provide VSMCs forming the tunica media of the descending and abdominal aorta as well as internal carotid, subclavian, and iliac arteries [39–41]. (4) The mesenchyme of the embryonic body wall – the so-called somatopleura – provides VSMCs forming the media of the limb arteries [15]. (5) The “visceral mesothelium” of the heart, lungs, and intraperitoneal organs provide VSMCs forming the media of the coronary arteries [42, 43], pulmonary arteries [44], and unpaired visceral branches of the abdominal aorta [45].
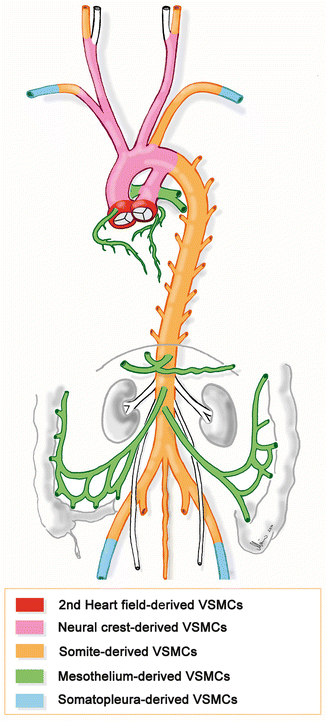
Fig. 1.4
Embryonic origin of vascular smooth muscle cells. Scheme showing the various embryonic origins of vascular smooth muscle cells forming the tunica media of the aorta and its main branches
Cellular and Molecular Mechanisms of Blood Vascular Development
In embryos, blood vascular development occurs a considerable time before development of lymphatics. Blood vessels develop by a number of different mechanisms involving the emergence of endothelial cells (ECs) from mesodermal angioblasts and hemangioblasts, followed by proliferation and coalescence of the cells to form a primary vascular plexus, as well as remodeling and differentiation of this plexus into maturing vascular networks, as outlined above. The initial phases of vascular development have been defined as vasculogenesis and angiogenesis [12, 13].
Vasculogenesis
Originally, the term “angioblast” was introduced by His [46], and the terms angiogenesis and vasculogenesis were used as synonyms that time with the general meaning of blood vessel development. The terms were then newly defined, unfortunately neglecting the existence of the second vascular system, the lymphatic vascular system. Thus, the blood vessels were the main focus, and vasculogenesis was defined as in situ differentiation of angioblasts or hemangioblasts into blood vessels [47, 48]. The growth of blood vessels from preexisting ones was called angiogenesis, but the two mechanisms cannot be clearly separated temporally and spatially [10, 49].
Already Sabin [17] proposed a subdivision of embryonic angiogenesis into three phases. Firstly, ECs are derived from precursor cells. These are either unipotent angioblasts (giving rise to endothelial cells) or bipotent hemangioblasts (giving rise to ECs and hematopoietic cells (HCs)) [18, 50]. The development of both ECs and HCs from a single precursor cell has not been shown unequivocally but was suggested because of their simultaneous development in blood islands. Evidence for the existence of hemangioblasts was provided mainly by in vitro studies in mice [51]. Here, it was shown that vascular endothelial growth factor receptor-2 (VEGFR-2)-positive embryonic stem (ES) cells are able to generate ECs as well as HCs. In accordance, VEGFR-2 null mice lack yolk sac blood islands and ECs [52]. In the chick, a single VEGFR-2+ cell can only give rise to either an EC or a HC colony [53]. Thereby, the emergence of ECs in vertebrates does not depend on normal mesoderm formation and can even be observed when gastrulation is prevented [54, 55], but, nevertheless, ECs and HCs are usually regarded as mesodermal cells. Secondly, endothelial cells are derived from precursor cells and simultaneously from ECs that are already integrated into the primary vascular plexus of the embryo. Thirdly, new endothelial cells solely emerge from already existing ones. This aspect has been challenged by the finding of circulating angioblasts in adults [56], which may, however, be of greater importance in pathologic angiogenesis than in embryonic angiogenesis.
There are two sources for blood vascular endothelial cells: mesodermal precursor cells (angioblasts and hemangioblasts) and proliferation-competent endothelial cells that exist as endothelial progenitor cells (EPCs) in vascular niches, which have not been defined unequivocally [57]. Angioblasts and EPCs possess high migratory potential. They migrate as single cells through the ECM or within the endothelial lining – preferentially against the blood stream [58, 59]. Angioblasts aggregate, flatten, and form tubes, so that the intercellular space becomes the lumen of the vessel [60]. However, intracellular lumen formation by confluence of vacuoles has also been described [61, 62], but live imaging studies in zebrafish again support the original view of lumen formation by expansion of the luminal cell membrane [63].
Angiogenesis
In earlier studies on embryonic angiogenesis, the term angiogenesis was used heterogeneously but was then commonly defined in a broad sense as the development of blood vessels from preexisting one, irrespective of the mechanisms involved in this process such as sprouting, intussusceptive vascular growth, splitting of vessels, and circumferential growth of vessels [13, 64]. In differentiated organisms, circumferential growth of arterioles and small arteries into larger arteries has been termed arteriogenesis [65]. In the early embryo, the initial (primitive) vascular plexus is made up of sinusoidal vessels lined solely by ECs. This early plexus has often been called “capillary plexus,” which is wrong because capillaries are organo-typically differentiated vessels with small lumen and often invested by a basal lamina and pericytes. Further growth of the initial vascular plexus is by integration of angioblasts and by a variety of mechanisms: mainly by sprouting and intussusceptive growth.
Sprouting
During sprouting the ECs extend long filopodia into their environment (Fig. 1.5). The leading cell of the sprout is called tip cell, while the more proximal cells are called stalk cells [66]. Tip cells and stalk cells communicate with each other as outlined below. The process of sprouting has been subdivided into sequential steps [67]: Firstly, there is local degradation of the vascular basement membrane by endothelial proteases [68]. This step, however, may not be of greatest importance in early embryos, since the extracellular matrix (ECM) is only just being produced, but is of great importance when ECs invade epithelial tissues such as the neural tube. Then, tip cells migrate toward angiogenic stimuli, which are presented as diffusible gradients, bound to the ECM or to neighboring cells. The short, intermediate, and long splice variants of VEGF fulfill these requirements: The short forms (121 amino acids) are highly diffusible, the intermediate forms (165aa) bind to the ECM, and the long forms (209aa) stick to the cell membrane of the secreting cell [63]. Then, lumen formation starts proximally in the sprout as a continuation of the lumen of the preexisting vessel. Lumen formation by confluence of intraendothelial vacuoles has been reported in intersomitic vascular sprouts of zebrafish embryos [69], but this finding was challenged as noted above. While tip cells usually do not proliferate, stalk cells undergo mitosis [70]. Proliferation studies with BrdU have shown that in the aorta of avian embryos, the labeling index of ECs declines from 30 % at stage 17 (HH) to 10 % at stage 29 (HH) [71]. Finally, adjacent sprouts anastomose, form loops, and become perfused with blood. The question how individual sprouts find each other has been resolved to some extent, when it was shown that angiogenic macrophages can regulate sprout anastomosis [72]. The Notch1 signaling pathway is active in these macrophages [73].
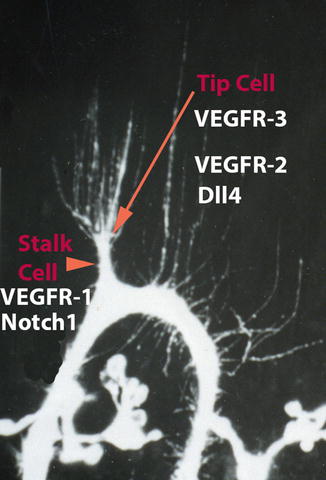
Fig. 1.5
Molecular characteristics of tip cells and stalk cells in vascular sprouts. Tip cells have long filopodia, are highly migratory, and express high levels of VEGFR-3, VEGFR-2, and Dll4. Stalk cells are highly proliferative, start forming a lumen, and express high levels of VEGFR-1 and Notch1 (Modified from Wilting et al. [49])
Interaction of Tip Cells and Stalk Cells
Angiogenesis proceeds by various mechanisms; however, most of our knowledge about the molecular control of this process has been discovered in sprouting angiogenesis. Sprouting is an important mechanism during the development of the central nervous system. There, the leading cell of the sprout, the “tip cell,” forms filopodia of up to 60 μm length [74]. The tip cell is a polarized migratory cell, which sits on a “stalk cell” that has proliferative potential and starts forming the lumen. The phenotypes of tip and stalk cells are transient and convertible. The Notch signaling pathway is high in stalk cells and low in tip cells (Fig. 1.5). The Notch ligand Dll4 is highly expressed in tip cells and suppresses tip cell behavior in stalk cells via Notch1 signaling. Notch1 upregulates VEGFR1 in stalk cells. This receptor acts as a trap for VEGF-A and thereby reduces tip cell characteristics. The main function of tip cells is the guidance of the sprout by filopodia. Tip cells express high levels of VEGFR-2 and VEGFR-3 and have low levels of Notch signaling activity. They migrate along a gradient of VEGF-A. Thereby, VEGF-A induces expression of Dll4 in tip cells, which by this means inhibit stalk cells from becoming a tip cell. Dll4-induced Notch signaling in stalk cells reduces expression of VEGFR-2 and VEGFR-3, but VEGFR-2 signaling is still sufficient to induce proliferation and lumen formation in stalk cells [66, 70].
Intussusceptive Microvascular Growth
The massive increase of the volume of developing organs by intercalation of cells is reflected by an overall expansion of the embryonic vascular plexus by a mechanism that has been called intussusceptive microvascular growth (IMG). The advantage of IMG is the possibility of a constant perfusion of the growing network. A detailed ultrastructural description of this process was provided by Burri and Tarek [75] and Patan et al. [76] for the developing lung and the avian chorioallantoic membrane. However, IMG can be observed in almost all parenchymal organs. In large sinusoidal capillaries, ECs form protrusions that “sprout” into the vascular lumen and make contact with the opposite luminal cell membrane. Intercellular junctions are formed and the endothelial cells become perforated centrally. Thereby, transluminal pillars are produced, which become invaded by mesenchymal cells and ECM. The pillars increase in diameter, thereby remodeling the sinusoidal vessel into a loop.
Circulating Endothelial Progenitor Cells
It was thought that the pool of angioblasts and hemangioblasts is completely used up during embryonic development, and in the adult new vessels are derived exclusively from preexisting ones. However, EPCs were identified in bone marrow and peripheral blood of adults on the basis of their expression of VEGFR-2 and CD34 [77]. However, these antigens are shared by angioblasts and hematopoietic cells (HCs). EPCs express VE-cadherin and AC133, which are lost once the cells differentiate into ECs [78]. Circulating EPCs seem to be of relevance during pathologic angiogenesis. They are recruited into sites of neovascularization in ischemic hind limbs, ischemic myocardium, injured corneas, cutaneous wounds, and tumor vessels [56, 77]. Data suggest that the circulating EPCs are of bone marrow origin and contribute to vasculogenesis mainly in the adult [79].
Remodeling of the Blood Vascular System: Arteriogenesis and Venogenesis
Soon after the formation of the initial vascular plexus, conduit vessels are formed. The first larger vessels are the aorta and the cardinal veins. In higher vertebrates and man, but not in fish, the aorta is initially formed from paired vessels, which fuse in the midline [80]. The specification of arterial and venous endothelium takes place in very early stages. The term arteriogenesis, however, has originally been proposed for the establishment of collateral arteries in ischemia [65]. The term “collateral formation” may be better suited to describe this process, and the terms arterio- and venogenesis seem to be more appropriate to describe the embryonic development of these vessel types.
The first markers that were found to differentiate between arterial and venous ECs were the juxtacrine signaling molecules EphrinB2 in arterial ECs and EphB4 in venous ECs [81]. Ephrins and Ehps are transmembrane molecules that bind to each other and induce signaling in both directions, forward and backward. EphrinB2 and EphB4 are expressed in arterial and venous ECs even before the onset of circulation but mainly appear to be functional during the remodeling of embryonic vascular networks [82]. The high plasticity of ECs has been demonstrated in grafting experiments that showed the capability of venous ECs to integrate into embryonic arteries during development, thereby adopting the arterial marker EphrinB2 [83]. Aberrant expression of the arterial marker EphrinB2 was observed in ECs of venous malformations [84]. Differentiation of embryonic arteries is accomplished by high expression of members of the Notch signaling pathway (Notch1, Notch4, Jag1, Jag2, Dll4, Hey2) and the upstream regulators sonic hedgehog (Shh) and VEGF-A.
Development of veins had been regarded as kind of a default pathway of vessel differentiation; however, the transcription factor COUP-TFII (nr2f2), an orphan nuclear receptor, was shown to induce venous specification. Disruption of its expression in murine ECs results in the upregulation of the arterial markers EphrinB2, Jag1, Notch1, and NP1 [80]. Development of veins is the prerequisite for the subsequent development of lymphatics.
Mechanisms of Lymphatic Vascular Development (Lymphangiogenesis)
Structure and Function of Lymphatics
Due to the pressure produced by the heart, 20–30 l of plasma leak each day from the blood vessels into the interstitium [85]. Ninety percent of the extravasated fluid is reabsorbed into the blood at the venous end of the capillaries and the post-capillary venules due to osmotic forces [86]. The remaining 10 % is taken up by initial lymphatics (lymphatic capillaries) and drained centripetally in lymphatic precollectors, collectors, and trunks [1]. Initial lymphatics are formed by only one cell type: lymphatic endothelial cells (LECs). A continuous basal lamina and pericytes are missing [87]. The diameter of initial lymphatics measures about 50 μm and is significantly larger than that of blood capillaries. LECs of initial lymphatics are interconnected by overlapping junctions, which, more appropriately, should be named “overlapping leaflets,” as they represent valves (first valve system), which allow the influx of fluid and hinder the efflux [88].
Precollectors measure about 100 μm in diameter. Their intermediate morphology still allows uptake of interstitial fluid, but occasionally they possess a muscular wall and a “second valve system.” This is made up of leaflets, formed by the intima in a comparable manner as in the veins. Next, collectors of up to 600 μm in width transfer lymph centrally. They possess numerous valves. Their wall is made up of three layers: intima, media, and adventitia. As a result of endogenous pacemaker cell activity and vegetative innervation, the nonstriated myocytes of the media contract spontaneously approximately 6–12 times per minute, which is of great importance for the lymph flow [2]. Collectors often drain the lymph over long distances (30–40 cm) into the first regional lymph node. Via lymphatic trunks and lymphatic ducts, such as the thoracic duct, the lymph is transported into the jugulo-subclavian junction [89].
Formation of Lymph Sacs and Primary Lymphatic Vessels
In the human, the heart starts pumping around the 21st developmental day, when blood vessels are already present [90]. Development of lymphatics starts considerably later. Lymph sacs, which are the first lymphatic anlagen detectable by routine histology, have been found in embryos of 10–14 mm total length, corresponding to developmental week 6 or 7 [91]. However, with specific markers LECs can be detected in earlier stages of development. Studies on mammalian embryos have shown that there are three paired and two unpaired lymph sacs, which will later give rise to primary lymph nodes [92]. The consecutive development of BECs and LECs has led to the hypothesis that LECs are derived from BECs, specifically from segments of adjacent veins [93]. This has been supported on the basis of the expression pattern of the homeobox transcription factor Prox1, which is indispensible for LEC development [94]. Direct proof for the development of LECs from venous ECs came from in vivo cell lineage tracing studies in zebrafish embryos. Cells delaminating from a parachordal vein assembled into the thoracic duct-homolog lymphatic vessel [95]. Budding and migration of lymphangioblasts from multiple vessels was observed during development of facial lymphatics using similar techniques in zebrafish [96]. In mice, too, the major source for the developing lymphatics is the embryonic veins [97]. Delamination from the endothelial lining and migration of mesenchymal lymphangioblasts, in combination with sprouting phenomena, then give rise to early lymphatic networks [98, 99]. In larger animals, e.g., chicken, indications for direct differentiation of LECs from mesenchymal lymphangioblasts have been found [100]. In the human, circulating lymphangioblasts were found to contribute to inflammation-induced lymphangiogenesis in patients suffering from kidney transplant rejection [101], but it is unknown if integration of mesenchymal lymphangioblasts is an important mechanisms of embryonic lymphangiogenesis in the human. Growth and expansion of lymphatics is preferentially along embryonic arteries, which obviously secret lymphangiogenic growth factors effectively, as well as chemokines, which orchestrate the patterning of lymphatic trunks [102].
Molecular Control of Lymphangiogenesis
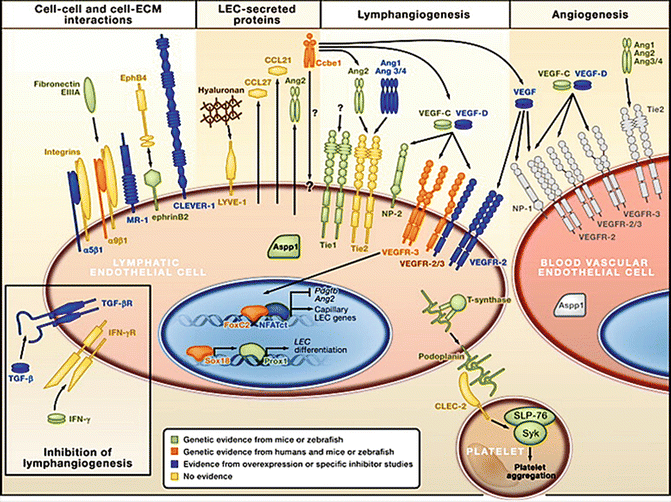
Commitment of LECs starts in specific segments of early embryonic veins, probably induced by adjacent mesenchymal cells and the regulation of retinoic acid concentrations [103]. Committed LECs express the transcription factor SOX18 [104], VEGFR-3, and the hyaluronan receptor Lyve-1 [105]. Upregulation of the homeobox transcription factor Prox1, which is a master regulator of LEC development, is a crucial step for the development of the lymphatic vascular network [94], and maintenance of Prox1 expression is found in lymphatics of aged humans [106]. Transcriptional profiling of LECs identified a large number of genes with high specificity for LECs, such as PDPN (podoplanin), RELN (reelin), WNT5A, and others [107–109]. High levels of VEGFR-3 expression were found in LECs of lymphangiomas [110], showing that fine tuning of the VEGF-C signaling is of major importance for tissue specific forming of lymphatics. This is underlined by the observation of lymphatic hyperplasia in mice after downregulation of soluble VEGFR-2 (esVEGFR-2), the endogenous inhibitor of VEGF-C signaling [111]. A soluble splicing variant of VEGFR-3 (esVEGFR-3) with anti-lymphangiogenic activity has been found recently [112]. The majority of familial cases of lymphedema seem to be due to mutations in the VEGFR-3 gene [113]. Molecules that regulate LEC development and the differentiation of lymphatic networks are shown in Fig. 1.6, taken from the review by Tammela and Alitalo [105]. For a detailed description of the molecular interactions, we recommend reading of this review. Not only cell-cell interactions but also the ECM controls lymphangiogenesis. Mutations of the CCBE1 protein (collagen and calcium-binding EGF domain-containing protein 1), which seems to control the composition of the ECM, cause lymphatic dysplasia in man, and functional analyses in animals have confirmed the importance of CCBE1 in embryonic lymphangiogenesis [114]. Important interactions of LECs with the ECM are mediated by integrin receptors, predominantly by integrin α9, which forms heterodimers with integrin β1. Inactivation of integrin α9 causes fatal chylothorax in mice [115].
 Hemangiomas of Infancy: Epidemiology
Epidemiology of Vascular Malformations
Hemangiomas of Infancy: Epidemiology
Epidemiology of Vascular Malformations
 Surgery of Hemangiomas
Surgery of Hemangiomas
 Dermatological Manifestations of Vascular Malformations
Dermatological Manifestations of Vascular Malformations
 Classification of Arteriovenous Malformation and Therapeutic Implication
Classification of Arteriovenous Malformation and Therapeutic Implication
 Vascular Malformations of the Limbs: Treatment of Venous and Arteriovenous Malformations
Vascular Malformations of the Limbs: Treatment of Venous and Arteriovenous Malformations




Related posts:
Hemangiomas of Infancy: Epidemiology
Epidemiology of Vascular Malformations
Surgery of Hemangiomas
Dermatological Manifestations of Vascular Malformations
Classification of Arteriovenous Malformation and Therapeutic Implication
Vascular Malformations of the Limbs: Treatment of Venous and Arteriovenous Malformations
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
