Pancreatic ductal adenocarcinomas account for approximately 85% of all pancreatic tumors. Recently, less common epithelial tumors have been increasingly identified as a result of the frequent use of computed tomography (CT). Solid pancreatic masses with atypical clinical presentations or unusual imaging characteristics may suggest an unusual etiology. At presentation, rare pancreatic conditions can be diagnostically challenging and a thorough differential diagnosis which includes, but is not limited to, pancreatic ductal adenocarcinoma can help to prevent errors in diagnosis. This chapter focuses on unusual solid tumors of the pancreas and discusses optimal diagnostic and therapeutic approaches for management.
Acinar cell carcinoma (ACC) is a rare pancreatic tumor accounting for less than 1% of pancreatic cancers. In contrast to pancreatic ductal adenocarcinoma, ACC arises from the acinar elements of the exocrine pancreas, not ductal epithelium. As a result, ACC often mimics the growth pattern of normal pancreatic acini and can produce digestive enzymes such as trypsin, chymotrypsin, and lipase. ACC occurs more commonly in men than women (2:1) and primarily affects individuals in the sixth and seventh decades of life. At presentation, approximately 50% of patients are asymptomatic at diagnosis; however, some patients may present with abdominal pain (45%) or weight loss (35%).1,2 Approximately 10% of patients with ACC present with a paraneoplastic syndrome caused by excessive pancreatic enzyme production, which is characterized by the presence of subcutaneous fat necrosis, bony infarcts, arthritis, and eosinophilia. No specific serum or plasma tests exist which are diagnostic for ACC, but serum lipase levels can be elevated in up to 25% of patients.1,3 Serum tumor markers such as carbohydrate antigen (CA) 19-9, alpha fetoprotein (AFP), and carcinoembryonic antigen (CEA) are variably expressed.3 In some patients, the combination of serum lipase and AFP can be quite helpful in assessing tumor burden in response to therapy.
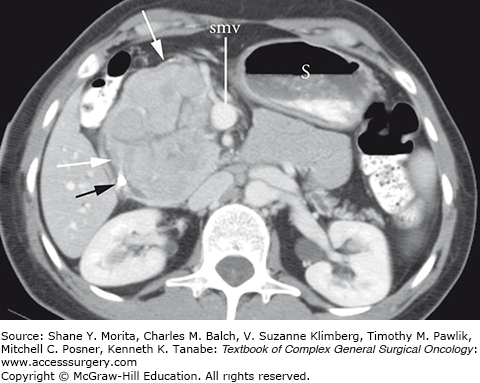
Acinar cell carcinomas tend to be large (mean diameter size of 7.1 cm2 to 10.6 cm2) and unifocal at presentation. The lesions can be entirely solid when small (and look identical to pancreatic adenocarcinoma on CT imaging), but larger tumors often outgrow their blood supply and develop central areas of necrosis. They have been reported to occur predominantly in the head of the pancreas (55%) but can also occur in the body and tail as well.4 Characteristic cross-sectional imaging findings include the presence of a large, exophytic, well-circumscribed mass with capsular enhancement but central hypodensity (Fig. 149-1). There may also be internal foci of calcifications.3 The radiographic differential diagnosis of ACC includes pancreatic ductal adenocarcinoma, pancreatic neuroendocrine tumor, solid pseudopapillary tumors, pancreatoblastoma, and mucinous cystic neoplasms.
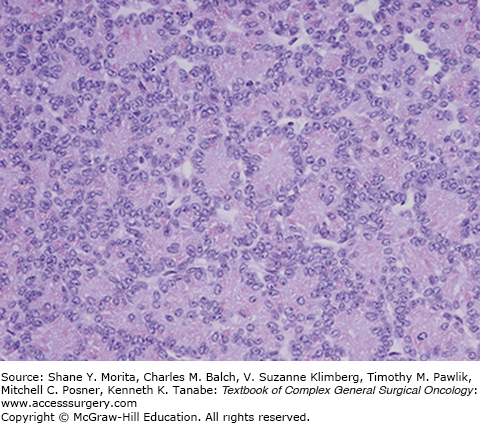
On histopathologic examination, pure ACCs have two predominant cellular patterns of growth: an acinar pattern consisting of cells growing in well-formed acini and a solid pattern characterized by sheets of cells that lack the prominent stromal component seen in pancreatic ductal adenocarcinoma.5 ACC cells have a uniform appearance with large, centrally located nucleoli with cytoplasm that is typically eosinophilic (Fig. 149-2). Classically, the majority of ACCs will have coarse granular apical cytoplasmic staining for trypsin or chymotrypsin.6 In contrast to the staining pattern of pancreatic ductal adenocarcinoma, ACCs generally stain negative for CEA and mucocarmine. Although fine-needle aspiration (FNA) biopsy can usually differentiate a pancreatic ductal adenocarcinoma from an ACC, the greater diagnostic dilemma is distinguishing between ACC and a well-differentiated pancreatic endocrine neoplasm and pancreatoblastoma. The diagnosis of ACC can be challenging owing to the morphological and immunophenotypical overlap that ACCs have with pancreatic neuroendocrine tumors. ACCs can have scattered neuroendocrine cells present in up to 40% of cells.5 To distinguish the neuroendocrine cells, additional immunohistochemistry for synaptophysin and chromogranin A may be informative (Table 149-1). When neuroendocrine cells comprise greater than 35% of the tumor, it qualifies as a mixed acinar-neuroendocrine carcinoma.7
| Feature | Type 1 | Type 2 |
|---|---|---|
| Structural involvement | Pancreatic ducts, lobules, artery, vein, and common bile duct | Mainly pancreatic ducts and intrapancreatic common bile duct |
| Immune infiltrate | Lymphoplasmacytic with eosinophils | Lymphoplasmacytic with neutrophils |
| Pancreatic ducts | Periductal inflammation without epithelial damage | Periductal inflammation with destruction of epithelium |
| Vessels | Obliterative phlebitis, rare arterial involvement | No phlebitis or arterial involvement |
| Peripancreatic tissue | May be included in the inflammatory process | Usually limited only to the pancreas |
| IgG4 immunostaining | Abundant (>10 cells/hpf) | Scant to absent |
Recently, whole exome sequencing of pancreatic neoplasms with acinar differentiation has been reported.8 Of the 21 ACCs that were sequenced, the average number of somatic mutations was 64 and no gene was mutated in over 30% of cancers. Several genes, including ATM, BRCA2, and PALB2, which have been associated with pancreatic ductal adenocarcinoma, were also identified in ACCs. However, key driver mutations, including KRAS, TP53, CDKN2A, and SMAD4, were infrequently mutated in ACCs. Interestingly, the genetic mutations associated with the major types of pancreatic cancer are now known to be relatively distinct: pancreatic ductal adenocarcinomas are characterized by mutations in SMAD4, TP53, KRAS, and CDKN2A; neuroendocrine tumors by mutations in MEN1, DAXX, ATRX, and the mTOR pathway; mucinous cystic neoplasms by mutations in RNF43; and intraductal papillary mucinous neoplasms by mutations in GNAS and RNF43. Adjunct sequencing may be valuable in tumors which are difficult to classify solely by histopathologic criteria.
Patients who have localized disease should undergo surgical resection. Although ACCs are generally large, they tend to be well circumscribed and are often amenable to complete surgical resection. In a review of the National Cancer Database, the 5-year survival rate of 865 patients who underwent surgical resection for ACC was 36.2%.9 Survivals from single institutional series are even more favorable, with median survivals reported as high as 57 months for patients with localized disease who undergo complete surgical resection.2,4 Adjuvant gemcitabine is often administered after complete resection, but ACCs have been reported to be less responsive to systemic chemotherapies as compared to pancreatic ductal adenocarcinoma. However, with the adoption of combination chemotherapy regimens, including chemotherapeutic regimens which utilize oxaliplatin and irinotecan, partial response rates in patients with metastatic disease have been reported in up to 30% of patients.9
Solid pseudopapillary tumors (SPTs) of the pancreas are rare neoplasms with low malignant potential and were first described in 1959.10 Historically, several other names have been associated with this tumor, including Frantz tumors, Hamoudi tumors, and papillary cystic neoplasm. It is estimated that SPTs represent up to 3% of all pancreatic tumors and 6% to 12% of pancreatic cystic neoplasms.11 SPTs are notable for their high prevalence among women, most commonly occurring in the third decade of life and earlier.12,13 The largest review of the literature included 718 patients over a 70-year time period.14 The authors observed that the prevalence of SPT is 10-fold higher in women than in men and affected predominantly younger individuals (mean age, 22 years; range, 2 to 85 years). More than 70% to 90% of patients with SPT present with symptoms, the most common being pain (45%) and/or an abdominal mass (34%).14 In the asymptomatic patient, tumors may also be discovered as a palpable mass on routine physical examination, as a mass seen on casual view of the abdomen (by a mother or father of an affected daughter), or as an incidental finding on imaging for an unrelated complaint. Serologic tests are often of little value with CA 19-9 elevated in 4.3%, amylase in 22.6%, and lipase in 29.3%.
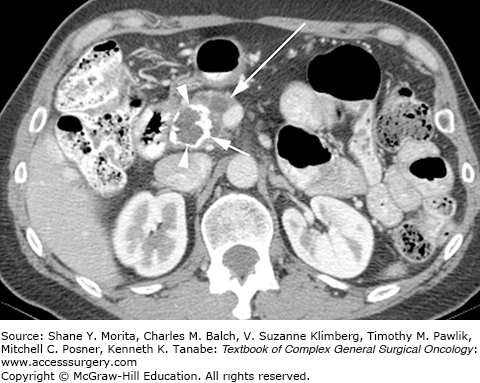
On CT imaging, SPTs are characteristically large, heterogeneously enhancing lesions with solid and cystic components, and they frequently demonstrate peripheral enhancement and central calcification (Fig. 149-3). These lesions can range from being completely cystic to completely solid.15 The cystic portion of SPTs is not a true cyst, but rather it has a cystic appearance which is secondary to necrotic degeneration of the primary tumor. As the solid papillary vascular stalks within the tumor slough and hemorrhage, central necrosis can occur resulting in cystic degeneration. Although SPTs can occur throughout the pancreas, they are perhaps slightly more common in the pancreatic tail and, when discovered, are generally large in size (mean diameter, 5.4 cm).15 Whether the clinical and radiographic features are prognostic of aggressive biologic behavior is controversial, largely because malignant behavior is so uncommon. In a recent review of 51 patients with SPT, no demographic, clinical, or CT imaging features were found to correlate with aggressive biology.15 In contrast, another series of 64 SPTs identified larger size (mean 10.5 vs. 5.2, p <0.001) as the sole prognostic factor for predicting disease recurrence.16 The radiographic differential diagnosis of a predominantly cystic SPT should include other cystic neoplasms including mucinous neoplasms or serous cystadenomas, and intraductal papillary mucinous neoplasms, as well as cystic degeneration of a typically solid neoplasm, such as a pancreatic neuroendocrine tumor or acinar cell cancer. In a young woman under the age of 30 (for example), SPT and pancreatic neuroendocrine tumor would be most likely; in a young woman under the age of 20, SPT would clearly be the most likely diagnosis. FNA biopsy may be useful when routine imaging is inconclusive and diagnostic uncertainty exists; however, because of the tumor’s largely necrotic composition, FNA biopsy can be nondiagnostic.
Solid pseudopapillary tumors have a characteristic microscopic appearance which include solid cellular hypervascular regions without gland formation, and the presence of branching papillary fronds with sheets and degenerative pseudopapillae.17 Cells have eosinophilic granules within the nuclei which are typically grooved. The immunophenotype demonstrates positive staining for neuron-specific enolase, CD10, and particularly atypical nuclear staining for beta-catenin, which is generally a cytoplasmic protein.17,18 Keratins, chromogranin, synaptophysin, and endocrine pancreatic enzymes are generally not expressed. SPTs are often stain positive for progesterone receptors, while estrogen receptor positivity is more variable.19 SPTs have also been reported to positively stain for α-methylacyl-coenzyme A racemase (AMCAR) in contrast to acinar and neuroendocrine tumors of the pancreas which do not.20 In 1996, the World Health Organization (WHO) further defined malignant SPTs as tumors with histologic characteristics of angioinvasion, perineural invasion, or extension into the surrounding pancreatic parenchyma.21 Loss of CD10 expression and high Ki-67 proliferative index has been associated with malignant SPTs.22,23
The molecular changes associated with the development of SPT have been well described and are distinct from the pattern of mutations seen in pancreatic ductal adenocarcinoma. As with ACC, the genetic profile associated with SPT is different from adenocarcinoma, most notably for an absence of KRAS and SMAD4 mutations. Almost all SPTs harbor alterations in the APC/beta-catenin pathway due to a mutation involving CTNNB1 (exon 3). Nuclear accumulation of beta-catenin has been described in 95% of SPTs and 74% of tumors overexpress cyclin D1, a downstream effector of beta-catenin.24 In addition, genes involved in the hedgehog and androgen receptor signaling pathways as well as genes involved in epithelial mesenchymal transition have been shown to be activated in SPT.25
Given the unpredictable but real metastatic potential of these tumors, surgical resection is recommended for all patients with localized SPT. Although these tumors may be extremely large and can invade critical vasculature, most lesions are usually amenable to complete resection. Pancreaticoduodenectomy or distal pancreatectomy can be performed with en bloc resection of involved adjacent organs when indicated. Complete margin-negative resection (R0) is associated with a 5-year survival rate of 95%.26 In a single-institution series of 24 patients, 17 of 19 patients underwent complete R0 resection. At a median follow-up of 8 years, no evidence of recurrent disease was observed in all patients who received an R0 resection.26 In another single-institution study of 37 patients with SPT, only 1 patient (3%) had a recurrence at median follow-up of 4.8 years.27
Given the excellent survival rates following surgical resection alone, adjuvant systemic therapy is not routinely utilized. If metastatic disease occurs, the most common sites include liver, mesentery, and peritoneum. Several series have reported long-term survival following metastasectomy.26 For unresectable metastatic disease, anecdotal case reports have suggested that gemcitabine-based chemotherapy may be successful in some patients.28,29
Related posts:
 Defining the Specialty of Surgical Oncology
Defining the Specialty of Surgical Oncology
 Medullary Thyroid Cancer
Landmark Clinical Trials that Impacted Surgical Management of Invasive and Noninvasive Breast Cancer
Multimodality Therapy of Gastric Cancer: Eastern Experience
Medullary Thyroid Cancer
Landmark Clinical Trials that Impacted Surgical Management of Invasive and Noninvasive Breast Cancer
Multimodality Therapy of Gastric Cancer: Eastern Experience
 Management of Hepatic Metastases from Neuroendocrine Tumors
Management of Hepatic Metastases from Neuroendocrine Tumors
 Skin Closure After Resection Of Skin Malignancies, Including Melanoma
Skin Closure After Resection Of Skin Malignancies, Including Melanoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


