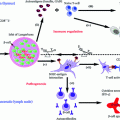
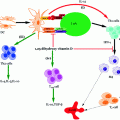
Fig. 4.1
Current therapeutic intervention strategies for T1D
Further progress in the field of type 1 diabetes patients has given rise to real-time continuous glucose monitoring (CGM). One of the first marketed product based on this technology was the GlucoWatch Biographer® (Cygnus Inc., Redwood City, CA) currently owned by Animas Corporation (Johnson & Johnson Companies, Milpitas, CA). Numerous other approved CGM devices include DexCom Seven® (DexCom, San Diego, CA), Paradigm RT®, and Freestyle Navigator (Abbott Diabetes Care, Alameda, California). These CGM devices are considered an adjuvant to self-monitoring of blood glucose in order to confirm information, such as hyper- and hypo-glycaemic events (Ellis et al. 2008; Renard 2002). CGMs with continuous subcutaneous insulin infusion (CSII) promoted research toward closed-loop systems. This system allows the delivery of insulin to the changing glucose levels in a real-time response. A closed-loop approach holds revolutionary power for glucose management. A closed-loop system, otherwise known as the artificial pancreas, comprises three parts, a CGM device to monitor glucose concentration, a titration algorithm to calculate the amount of insulin to be delivered, and an insulin pump to deliver the computed dosage of insulin (Fig. 4.2). Primarily, three types of closed-loop systems are defined as follows: (1) a sc–sc system with a subcutaneous (sc) glucose sensing and sc insulin delivery, (2) an iv–ip with an iv sensing and intraperitonial (ip) delivery system, and (3) an iv–iv system with iv sensing and iv delivery (Renard 2015; Renard and Schaepelynck 2007). Given the minimal invasion and pervasive usage of external insulin pumps, the sc–sc approach had the potential to achieve a widespread application. Even so it may not be compatible with a fully closed-loop system due to considerable delays prohibiting safe and timely control of large or rapidly absorbed meals. Iv–iv approach is used in particular situations such as in the critically ill patients, comatose, or for research investigations. The shortcoming of this approach is its invasiveness which jeopardizes patient to infections and clotting problems (Hovorka 2008). Therefore, the uses of closed-loop systems are particularly appealing, and these include glucose monitoring with an insulin pump that is encompassed within an artificial pancreas (Powers 2008; Hovorka 2005). These devices not only closely monitor the blood glucose level but also deliver insulin in body in a controlled way. They aim to reduce the risk of hypoglycaemia while achieving tight control of glucose (Elleri et al. 2011). Clinical testing on closed-loop systems so far presented positive results with regards to safety and efficacy. It was found that overnight closed-loop delivery of insulin improved overnight glycaemic control and less risk of nocturnal hypoglycaemia compared to patients treated with conventional insulin pump (Breton et al. 2012; Buckingham et al. 2010). One of the novel techniques for continuous immunosensors is weak affinity chromatography which can sense molecules with fast off-rate and disassociation constant (Kd) in ≈ mM range necessary for detection of dynamic change in analyte concentration in the blood. Monoclonal Ab (3F1E8-A2) has been evaluated for their future potential in glucose sensor and was found to be specific for glucose molecule compared to other monosaccharides with a Kd = 18.8 ± 2.6 mM (Engström et al. 2008). The major stepping stone in the glucose sensor future will be its clinical validation for diabetes control.
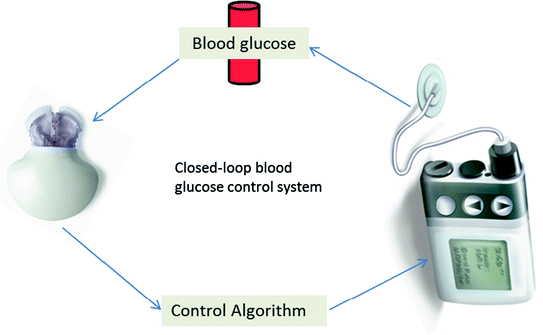
Fig. 4.2
Key elements involved in the restoration of closed-loop blood glucose control system in type 1 diabetes
4.3 β-Cell Replacement: Transplantation Versus Regeneration
4.3.1 Islet and Pancreas Transplantation
An alternative therapeutic option to conventional insulin replacement therapy is based on islet or pancreatic transplantation and replacement of β-cells with insulin-producing cells obtained from sources other than islet cells. The objective behind replacement of β-cells either with whole pancreas or islet transplantation is to attain long-term insulin independence, and the associated benefits which include better quality of life and prevention of secondary complications such as nephropathy, retinopathy, and vasculopathy (Sutherland et al. 2004). Pancreatic transplantation is an established procedure using the Edmonton protocol (Shapiro et al. 2006). Primary results revealed poor graft survival rates of less than 5 % after 6 months (Kelly et al. 1968) although simultaneous transplantation of a whole pancreas and kidney, albeit highly invasive, has delivered significant improvements in patient response and prognosis (Humar et al. 2000).
Comparatively, islet transplantation seems a more promising approach for β-cell replacement since it is a noninvasive procedure that involves percutaneous cannulation of the portal vein. Furthermore, islet grafts can be stored in culture tissue or can be cryopreserved (Ryan et al. 2001; Nanji and Shapiro 2006). Despite advancements in this field, a 5-year follow-up study showed that a mere 10 % of the tested population remained insulin independent for a median period of 15 months after transplantation (Ryan et al. 2005). In fact, a major setback to the approach is the difficulty to protect the transplanted islets from host immune attack (Humar et al. 2000).
However, advancements in the techniques to procure islets from cadaveric donors, less toxic immunosuppression regimens, and techniques such as xenotransplantation which makes use of islets from sources other than humans (e.g., porcine) will no doubt provide the much required edge to islet transplantation as a choice of therapy in the imminent future. Immune reactions against xenotransplant tissue are one of the lingering hurdles associated with this approach. Nevertheless, immunosuppression alongside innovative techniques such as islet encapsulation in alginate microcapsules, which protect the islets from T-cell mediated responses, have shown much promise to provide breakthroughs leading to greater immune tolerance of xenotransplanted tissues (Zimmermann et al. 2005). In addition to immune tolerance following xenotransplantation, there also exists a very real risk of porcine endogenous retrovirus transmission to the host (Limbert et al. 2008) and hitherto data available to establish the prevalence of infection in transplanted patients remain inconclusive, questioning the widespread use of this approach.
4.3.2 Islet Regeneration
As described above, development of T1D occurs as a consequence of loss of functional β-cells. With this in mind, the primary goal of therapeutic interventions to treat T1D is to restore β-cell mass, in sufficient amount to maintain euglycemia. Encouraging results with clinical islet transplantation trials bolstered a novel idea to exploit the regeneration capacity of the pancreas and other related tissues (SoRelle and Naziruddin 2011). It is well established that in healthy individuals, lost β-cells are constantly replenished in vivo through β-cell regeneration; however, further progress in this area has been delayed due to a poor understanding of the mechanisms driving the replenishment process. Some scientists argue that it happens via β–cell neogenesis while, according to other experts, self–replication of β-cells takes place. As evidence, postnatal origin of β-cells using a genetic lineage method in pancreatectomized mice was investigated. The study demonstrated that mature β-cells possess the capacity to proliferate; the replacement of lost β-cells is mainly due to replication of the existing cell mass (Dor et al. 2004), whereas β-cell neogenesis occurs through progenitor cell activation and/or transdifferentiation of mature fully differentiated cells, as a means of β-cell mass expansion (Lipsett et al. 2006).
Current avenues under investigation for islet cell expansion include ex vivo expansion, which require the addition of pharmacological agents, and encouraging results have been reported in both in vivo animal and in vitro human models (Beattie et al. 1997). Various hormones and growth factors are also known to increase β-cell mass; these include gastrin, glucagon-like peptide (GLP-1), epidermal growth factor (EGF), and islet neogenesis-associated protein (INGAP). Regrettably, over the course of time these differentiated cells lose their capacity to secrete insulin, possibly due to aging. Till date, several combination therapies with EGF + gastrin and GLP-1 + gastrin have shown promising results in β-cell expansion (Brand et al. 2002; Suarez-Pinzon et al. 2008). Unfortunately, none of the studies have focused its application specifically on T1D.
Another pharmaceutically synthesized ligand under investigation is INGAP (INGAP104−118), first identified in hamster model of islet expansion. INGAP administration was found to increase functional β-cell mass in animals and therapeutically reverse hyperglycemia in streptozotocin-induced (STZ) diabetic models. The authors demonstrated that this pentadecapeptide is capable of inducing duct and acinar cell-associated islet neogenesis, which in turn was associated with PDX-1 expression. PDX-1 is considered an important transcription factor required in normal islet development (Rosenberg et al. 2004; Jamal et al. 2005).
4.3.3 Stem Cell-Derived β-Cell Replacement
In order to address critical shortfalls of islet donors, stem cells appear to be an excellent alternative, being an unlimited source of β-cells, with the added potential of high differentiation and tissue regeneration. The most commonly used type of stem cells are embryonic (ESC) and autologous adult stem cells (ASC). ESC are derived from the inner mass of the mammalian blastocyst and have shown to be an exceptionally versatile source of stem cells with immense pluripotency and proliferative capacity (Thomson et al. 1998; Zulewski 2006). Soria et al. amongst the first teams successfully demonstrated that ESC are capable of maintaining a stable glucose response in streptozotocin (STZ)-induced diabetic mice in vivo (Soria et al. 2000). STZ is toxic to the insulin-producing β-cells in mammals, and therefore it is restricted to the engineering of animal model for T1D. The above study is analogous to recent studies by Clark et al. (2007) and Kroon et al. (2008), reporting the assessment of pancreatic endoderm derived from human embryonic germ (EG) cells and human ESC, respectively, with efficient production of glucose-responsive endocrine cells in vivo (in engrafted mice). Following transplantation into mice, differentiated cells have been identified by co-expression of transcription factors such as PDX1, FOXA2, SOX17, HNF6, and NKX6-1, marking a definitive pancreatic endoderm. Approximately, 60–70 % of differentiated ESC produced the same level of glucose as compared to other studies in mice implanted with ~3000–5000 adult human pancreatic islets (D’Amour et al. 2006). However, attempts to assess the amount of insulin generated by ESC have yielded disappointing results, with differentiated ESC under any specific culture condition producing only 1.6 % of the total insulin secreted by that of normal β-cells (Devaskar et al. 1994). Furthermore, despite the ESC capacity to differentiate into insulin-producing cells, there is a remote possibility that insulin-positive cells in ESC cultures may be a product of insulin uptake from the medium rather than an endogenous outcome. In addition, this approach is circumscribed, due to the ethical issues concerning the use of human ESC and their carcinogenic potential, low efficiency, and high cost.
ASC are further classified into hematopoietic stem cells (HSC) and mesenchymal stem cells (MSC), primarily obtained from the bone marrow (BM) with transdifferentiation plasticity, thus capturing great interest as a future therapy for T1D and other metabolic diseases. The promising role of MSC relies on two main aspects: first, they are multipotent and possess certain essential growth factors, which support tissue regeneration. Second, they present a broad range of immunomodulatory properties by interaction with a wide range of immune cells and hence this property can be harnessed to limit allograft rejection post-transplantation (Brusko 2009; Vija et al. 2009). Sun et al. demonstrated the in vitro differentiation of MSC into insulin-producing cells obtained from diabetic patients (Sun et al. 2007). The study further suggested that MSC could be used as a source of insulin, thereby reducing the risk of graft rejection. Voltarelli et al. conducted the first clinical trial in an attempt to explore the autologous potential of non-myeloablative HSC transplanted in T1D patients. A high-dose immunosuppression regimen was followed after post-transplantation (Voltarelli et al. 2007). After a mean follow-up period of approximately 9 months, increased C-peptide levels were seen in ~95 % of the patients, with adequate glycaemic control over a considerable period of time thereafter and with acceptable adverse effects reported.
As discussed earlier, MSC modulate the immunogenic properties of immune cells. In order to investigate this aspect, human-derived MSC (hMSC) were co-cultured with different subpopulations of immune cells in vitro, most notably T-cells. Here, hMSC influenced cytokine secretion levels with reduced levels of pro-inflammatory cytokines (TNF-α, IFN-γ, interleukin (IL)-6, and IL-17) and elevated levels of anti-inflammatory cytokines (IL-10, IL-4, and prostaglandin E2) (Aggarwal and Pittenger 2005; Vija et al. 2009; Ben-Ami et al. 2011). It has been corroborated that these mediators do indeed play a crucial role in the MSC-mediated immunosuppression. Another possible role of hMSC is to increase the Treg cells level, which in turn down regulates T-lymphocyte proliferation and consequently suppresses autoimmunity (Aggarwal and Pittenger 2005; Augello et al. 2005). Furthermore, the question remains whether the effect exerted by MSC is due to the release of cytokines or due to cell-to-cell contact between MSC and T-cells (Werdelin et al. 1998; Aggarwal and Pittenger 2005; Augello et al. 2005; Selmani et al. 2008). There are several conflicting studies in the literature, which lead to the conclusion that there is most likely a range of mechanisms contributing to MSC-mediated immunomodulation.
At present, several human clinical trials deploying MSC-based therapies are underway. Osiris therapeutics is conducting a phase II study (Clinical trial no. NCT00690066) using ex vivo cultured ASC (Prochymal®) for treatment in recently identified, early onset T1D patients. Preclinical studies thus far have not shown any adverse effect of the allogenic MSC, and whether this remains the case in a clinical setting will become apparent as clinical trial data become available. To circumvent anticipated adverse events from allogenic MSC, much effort has been invested toward encapsulation techniques that protect the transplanted cells. Encaptra®, an advanced drug delivery system, is under development by Viacyte, Inc. Encaptra® consists of islet-like structures that release insulin in response to changes in physiological glucose levels in vivo. It is manufactured from FDA compliant implant grade materials, and is a SC implantation device designed with the goal to exclude the need for continuous immunosuppression.
Cell-based therapies are touching upon new undiscovered horizons and hence an increased understanding of the various factors contributing to β-cell mass dynamics is warranted. This will enable researchers to experiment with new approaches to induce β-cell mass expansion and promote their survival. The current knowledge of the field points toward seeking an absolute therapy for T1D, and the approaches outlined above, namely, islet formation, promotion of cell survival, and long-term maintenance of function, as well as the prevention of recurrent autoimmunity, should be examined simultaneously albeit judiciously.
4.4 Immune Intervention Strategies
From T1D pathophysiology, it is apparent that the primary physical indicator of β-cell destruction presents itself as insulitis, accompanied by the infiltration of CD4+ and CD8+ autoreactive cells. Based on murine studies, it is believed that during ongoing insulitis, ~80–90 % β-cells are damaged even before clinical diagnosis of T1D (Sreenan et al. 1999); this appears a dubious statement and only recently a meta-analysis study was carried out to gauge its validity Klinke (Klinke 2008). The conclusions drawn from the study paint a more detailed picture, indicating that the level of β-cell destruction varies with age and furthermore and depends on the insulin-producing capacity, which is proportional to β-cell mass, with body weight also playing a key role (Klinke 2008). Bearing these factors in mind, current immunotherapeutic approaches embody several innovative strategies, which include antibodies, antigens, and cytokines in individual- or cocktail-based therapies, and the section below provides an in-depth account of state-of-the-art approaches being pursued as treatment options for T1D.
4.4.1 Nonantigen-Based Intervention Strategies
Nonantigen-based intervention is primarily focused on the prevention of autoimmunity by modulating or silencing the immune response in the absence of negative effects on Tregs cells.
4.4.1.1 Immunosuppression
Immunosuppression is amongst the earliest reported intervention strategies used at the onset of T1D (see Table 4.1 for comprehensive list of immunosuppressive technologies); however, it has proven of limited use with only partial or in some cases no remission of T1D.
Table 4.1
Immunosuppressive agents in T1D and their mechanism of action
Immunosuppressive method/agents and MOA | References |
|---|---|
Plasmapheresis  • Partial or no success • Drawback: Need for immunosuppressive drugs, and a painful procedure hence not advisable in children | |
Cyclosporine (CsA) 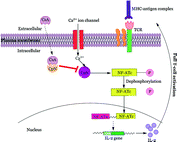 • Maintains C-peptide and reduces HbA1c levels • Diabetes French study: 46 % remission rate in treatment group with few (minor) adverse effects reported • Drawbacks: Dose dependence, long-term malignancy, not optimal for children | |
Nicotinamide (vitamin D)  • High dose prevents or delays onset of T1D • DENIS and ENDIT studies failed to show any significant improvement in diabetes risk; No adverse effects observed |
4.4.1.2 Antibody-Based Immunotherapy
Given that immunosuppressive intervention in T1D to date has failed to live up to its promise, it remains the challenge to find therapies that are not only highly efficacious but also safe when translated into humans, where exposure to exogenous antigens can be altogether avoided. The solution to this conundrum may well lie in the employment of a targeted approach, whereby monoclonal antibodies (mAb) can lead the way by directing Teff cells, with the former also acting as biological immunomodulators (Li 2011). Some of the mAbs specifically designed for this purpose include anti-CD3, -CD20, -CD25, anti-thymocyte globulin (ATG), and CTLA-4, which target immune Teff cells and many are presently under investigation in a range of T1D clinical trials.
T-Cell Targeting Antibodies
Anti-CD3 Antibody (Otelixizumab and Teplizumab)
Orthoclone-OKT3® (Muromonab-CD3 antibody) was the first US-FDA approved murine-derived anti-CD3 mAb, used in humans to prevent acute rejection post-organ transplantation. Preclinical studies in NOD mice showed that treatment with OKT3® resulted in potent reversal of T1D and successfully prevented immune responses toward the transplanted syngeneic islets (Herold et al. 1992; Chatenoud et al. 1994; Chatenoud and Bluestone 2007). As the name suggests, these mAbs are targeted to CD3 present on the surface of T-cells, which are necessary for T-cell activation. The immunomodulatory mechanism of CD3 mAb is suggested to act in two phases: phase I is attributed to T-cell depletion (preferentially Teff cells) and activation of cells that secrete a range of cytokines (IL-10, TNF-α, IFN-γ). Phase ΙΙ is proposed to have long-lasting effects, which involve induction of Treg cells with enhancement of CD25+ Forkhead box protein 3 (Foxp3) population, which is further dependent on transforming growth factor beta (TGF-β) -β) (Waldron-Lynch and Herold 2011). These postulated mechanisms render anti-CD3 a potential therapeutic agent; however, its side-effect profile includes transient cytokine syndrome which causes fever, hypotension, and arthralgia. The side effects were supposedly linked to activation of T-cells due to binding of murine Fc portion (fragment crystallizable region of antibody) of the antibody with the Fc receptors (FcR) present on human cells (Abramowicz et al. 1989; Chatenoud et al. 1989).
In order to mitigate the effects of anti-CD3 mitogenic potential humanized, non-Fc binding anti-CD3 mAbs were engineered (Table 4.2). Teplizumab (humanized non-Fc-engineered mAb, also known as MGA031, hOKT3γ1, and Ala-Ala) and Otelixizumab (aglycosylated chimeric/humanized non-Fc binding mAb, also known as TRX4 and chAgCD3) have shown significant improvement in terms of safety due to their fully humanized profile, minimizing the potential for immunogenicity associated with the differences in mAb isotype between humans and mice (Keymeulen et al. 2005). Nonetheless, adverse events have been reported with anti-CD3, which include flu-like symptoms due to transient cytokine release as well as increased incidences of EBV infection following a single course of anti-CD3; this was, however, self-limiting with EBV incidence subsiding 1–3 weeks post-treatment initiation (Keymeulen et al. 2010a, b).
Table 4.2
Outcome of various anti-CD3 antibody-related clinical trials
Study title | Details | Results | Reference |
|---|---|---|---|
Teplizumab mAb (Anti-CD3 mAb/hOKT3γ1/Ala–Ala) | |||
Autoimmunity-blocking antibody for tolerance in recently diagnosed type 1 diabetes (AbATE) | • Phase II; n = 81 (age range: 8–30 year) • Duration post-diagnosis: 8 weeks • Status: active • ClinicalTrials.gov Identifier: NCT00129259 | Ongoing | Herold et al. (2009) |
Protégé Study-clinical trial of MGA031 in children and adults with recent onset type 1 diabetes mellitus | • Phase II/III; n = 516 (age range: 8–35 year) • Duration post-diagnosis: 12 weeks • Status: complete • ClinicalTrials.gov Identifier: NCT00385697 | β-cell preservation (detectable C-peptide level), lower insulin requirement | Sherry et al. (2011) |
Anti-CD3 mAb treatment of recent onset type 1 diabetes (Delay) | • Phase II; n = 60 (age range: 8–30 year) • Duration post-diagnosis: 4–12 months • Status: complete • ClinicalTrials.gov Identifier: NCT00378508 | Ongoing | |
Teplizumab for Prevention of Type 1 Diabetes In Relatives “At-Risk” | • Phase II; n = 140–170 (age range: 8–45 year) • Subjects with high risk • Status: recruiting • ClinicalTrials.gov Identifier: NCT01030861 | Ongoing | |
Otelixizumab mAb (Anti-CD3 mAb/TRX4/chAglyCD3) | |||
Extension of Phase II therapeutic trial with a humanized non-mitogenic CD3 (ChAgly CD3) monoclonal antibody in recently diagnosed Type 1 diabetic patients | • Phase II; n = 80 (age range: 12–45 year) • Duration post-diagnosis: 4–12 months • ClinicalTrials.gov Identifier: NCT00627146 | Reduced insulin requirement over a period of 2 years, depending on their age and initial residual beta cell function | |
Trial of Otelixizumab for adults with newly diagnosed type 1 diabetes mellitus (autoimmune): DEFEND-1 | • Phase III; n = 272 (age range: 12–45 year) • Duration post-diagnosis: 90 days • Status: complete • ClinicalTrials.gov Identifier: NCT00678886 | Failed to meet primary end point of change in C-peptide level at 12 months | |
Trial of Otelixizumab for adolescents and adults with Newly diagnosed type 1 diabetes mellitus (autoimmune): DEFEND-2 | • Phase III; n = 396 (age range: 12–17 year) • Duration post-diagnosis: 90 days • Status: active but not recruiting • ClinicalTrials.gov Identifier: NCT01123083 | Suspended pending reviews for DEFEND-1 | |
Future developments of anti-CD3 antibodies should continue to focus on improving the safety along with increasing the efficacy. An additional factor to be considered is the stage of disease at the time of drug administration, as more promising results were observed in preclinical models when these agents were introduced in the earliest stages of the disease. Five independent large-scale clinical trials presently underway (phase II/III) have reported some encouraging outcomes (see the details in Table 4.2). The best response to therapy was noted in patients with a greater β-cell mass prior to initiating therapy; this implies that anti-CD3 therapy, if combined with agents that improve β-cell regeneration such as GLP-1 or other mimetics, may prove to be a superlative approach (Sherry 2007). One such attempt at this dual approach was reported with a combination of anti-CD3 antibody with intranasal proinsulin peptide, yielding encouraging results (Bresson et al. 2006).
Anti-CD25 (Daclizumab)
Another promising immunomodulatory antibody is anti-CD25 (Daclizumab), a humanized mAb which binds CD25, the α-subunit of the IL-2 receptor expressed on T-cells, and by doing so it inhibits the activation of T-cells. Daclizumab has been applied in autoimmune conditions such as multiple sclerosis (MS) and uveitis (Bielekova et al. 2009) although studies showing its effect in T1D monotherapy are absent. A recent study in diabetes-resistant bio-breeding (DR–BB) rat model demonstrated the synergistic effect of Daclizumab and Mycophenolate mofetil (MMF) (with MFF having a cytotoxic effect on autoreactive T-cells) (Ugrasbul et al. 2008). Disappointingly, a recent phase III clinical study using the same combination therapy failed to preserve β-cell function in new-onset T1D patients (Gottlieb et al. 2010). Moreover, although nonsignificant, a higher incidence of side effects was observed with the dual therapy, when compared to the placebo group.
Anti-thymocyte Globulin—Thymoglobulin®
Thymoglobulin® (anti-thymocyte globulin/ATG) is a purified γ-immune globulin raised in rabbits and horses via immunization against human thymocyte. This immunosuppressive agent contains cytotoxic antibodies directed against antigens expressed on human T-cells (Simon et al. 2008). Currently, rabbit-derived ATG is licensed as a treatment for acute rejection in renal transplant patients across Europe and USA. It is also indicated in therapies such as graft host disease (GVHD), aplastic anemia, and cancer. It was reported that in NOD mice, administration of murine ATG prevented progression of new-onset diabetes and induced long-term tolerance. The mechanisms involved are similar to anti-CD3, where first transient but substantial increases in cytokine release (TNF, IFN-α, IL-2, IL-3, and IL-6) (Ferran et al. 1991) are observed. Next, there is transient deletion of T-cells (Teff) with increased frequency of CD4+CD25+ Treg cells, although the levels of Tregs cells were found to return to baseline after 30 days of treatment with ATG. In vitro and in vivo data support another plausible explanation, i.e., alteration of DC cells causing reduced expression of CD8+ expression along with increased IL-10 production (K. Womer, unpublished study). Nonetheless, optimal outcomes with ATG therapy are dictated not only by dose, but also by the time from diagnosis and when therapy is initiated, with maximum efficacy seen when administration occurs after recent onset or late in the prediabetic phase (at 12 weeks of age). In complete contradiction to these findings, there is now evidence suggesting that the effect of ATG antibodies is dependent on the animal model employed (Bresson and Von Herrath 2011). This suggests that, before making any broad conclusion on the applicability of antiglobulin therapy, it is essential to assess it in various models of the disease, other than the most popular ones (e.g., NOD mice and BB rats) models.
Transitioning from trials in animals to those in humans, there are reports of a combination therapy, using ATG alongside prednisone. Positive results with lowered HbA1c levels and decreased insulin requirement per day were observed over several months. However, adverse effects of ATG therapy were also noted; these included cytokine release syndrome (leading to symptoms like fever and serum sickness) and even thrombocytopenia, which precluded its further use (Saudek et al. 2004). Currently, a trial entitled Study of Thymoglobulin to Arrest Newly Diagnosed Type 1 Diabetes (START) (Clinical trial ID: NCT00515099) aims to determine whether ATG treatment can halt the progression of newly diagnosed T1D when given within 12 weeks of disease diagnosis. Further, a combination trial in phase I/II has commenced using Thymoglobulin® and granulocyte colony-stimulating factors (GCSF) (Neulasta®) to identify their safety and potential to preserve insulin production. This combination therapy has shown durable remission of T1D in NOD mice due to the induction of Treg cells (Parker et al. 2009).
Cytotoxic T-Lymphocyte Antigen 4 Immunoglobulin (CTLA4-Ig)
The fusion immunoglobulin CTLA4-Ig (Abatacept®) acts on a number of costimulatory molecules (e.g., CD80 (aka B7-1) and CD86 (aka B7-2)) present on APCs (Fig. 4.2), functioning as a negative regulator of T-cell response (Perrin et al. 1995; Arima et al. 1996). T-cells require a secondary signal, in addition to the primary signal transmitted via MHC/TCR interaction, for their full activation. Hence, CTLA4-Ig acts by blocking CD28/B7 interaction (essential for the activation of T lymphocytes), leading to anergy (for further explanation, refer to the previous Sect. 3, Genetic susceptibility). Recently, blockade of the CD28/B7 costimulatory pathways has gained much attention, since it demonstrated to halt the progression of ongoing autoimmunity (Lenschow et al. 1992). An additional study reported that a single dose of CTLA4-Ig resulted in long-term graft survival after allogenic transplantation; this was marked by increases in cytokine IL-4 and IL-10, both of which are characteristic of Th2 subtype population. Separately, they were also found to induce production of TGF-β, a key regulatory cytokine involved in the immune regulation, thus supporting their role in the prevention of autoimmunity (Salomon et al. 2000; Salomon and Bluestone 2001; Tang et al. 2003). Furthermore, these immunoglobulins induce tolerance in allergenic islet transplantation by indolamine 2, 3-dioxygenase (IDO) mechanism (Fig. 4.3). IDO is a tryptophan degrading enzyme secreted by specific DCs, and hence it controls T-cell activation (Alexander et al. 2002). However, in certain cases, opposite to the CTLA-4 preventive action, it is found to augment autoimmune conditions. This could be due to (a) blocking of CTLA-4, which is a negative regulator of T-cell activation and (b) direct impact on the Tregs whose survival and differentiation depend on CD28 (Lenschow et al. 1996; Rigby et al. 2008). Hence, their blockade further results in exacerbation of T1D. The overall discussion so far seems to show that blocking CD28/B7 pathway on one hand may provide benefit in preventing or at least delaying the autoimmune disorder. On the other hand, its complex involvement in the maintenance of Tregs somehow makes its utility as an immune-modulating agent difficult. Other potential issues such as toxicity, time dependence, and site of administration need critical evaluation and to this end phase II clinical trials are underway using CTLA4-Ig (Abatacept®) in cases of new-onset T1D (Orban et al. 2011) and LEA29Y (Belatacept®) in islet transplantation (Chatenoud 2011).
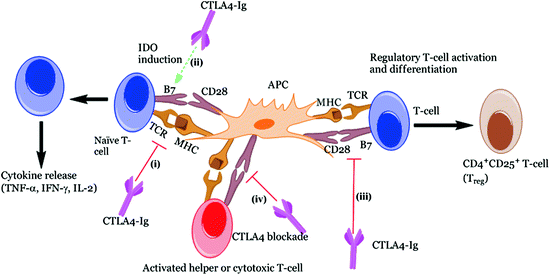
Fig. 4.3
Different roles of CTLA4-Ig implicated in immune response. CTLA4-Ig exerts its action via several different mechanisms: (i) CTLA4-Ig blocks the CD28 interaction with B7 owing to its high affinity for B7 co-receptor and results in blocking of T-cell activation or anergy, (ii) Induction of IDO following interaction of CTLA4-Ig with B7 which prevents T-cell activation, (iii) Immune suppression effect due to the blockade of CD28 resulting in reduction of Th2 cell differentiation, i.e., Treg cells and finally, (iv) Inhibits the CTLA-4 binding with B7 ligands necessary for CTL activation which are essential to control Teff cell function
B-Cell Targeting Antibody
Literature abounds with illustration of the clear effector role of T-cells in autoimmune T1D and hence most of the therapeutic studies target T-cells for treatment purposes. From our previous discussion, we know that NOD mice demonstrate a plausible role of B-cells as APC in T1D pathogenesis, more than just producing antibodies against autoantigens. This somehow indicates that therapies such as anti-CD20 and anti-CD25, which target B-cells, may have a positive role in the disease remission. In fact, anti-CD20 mAb treatment in NOD mice has shown depletion of CD20 B-cells, which prevented T1D in more than 60 % of tested mice population due to their predominant interference with CD4+ T-cell activation (Serreze and Silveira 2003).
Anti-CD20 (Rituximab)
Rituximab is a chimeric mAb targeted against CD20 transmembrane receptors, found primarily on the surface of B-cells (Looney 2005; Martin and Chan 2006). Rituximab was first introduced in 1997 and is the first therapeutic mAb, approved by the European Medical Agency (EMEA) and US-FDA, for the treatment of B-cell non-Hodgkin’s lymphoma (NHL). It is additionally used in other therapies such as rheumatoid arthritis, systemic lupus erythematosus (SLE), and multiple sclerosis (MS) (Pescovitz et al. 2011). A key advantage of rituximab is its specificity for CD20 molecules compared to other treatment strategies, which prevents undesired side effects. A clinical trial sponsored by TrialNet included 87 patients (age range: 8–40 years) with newly diagnosed T1D, who received 4 weekly infusions of rituximab. The study reported that, after 1 year, stimulated C-peptide level was found to be 20 % higher in the rituximab-treated group compared to the placebo group. Secondary treatment responses included lowered HbA1c level and lowered daily doses of insulin with no increase in infection or neutropenia observed. Unfortunately, the C-peptide response was short lived with maximal drug effect being apparent only in the early months of treatment (Pescovitz et al. 2009). This suggests that rituximab may not be an optimal approach for T1D treatment but certainly it provides a proof-of-concept that B-cell depleting agents are a perfectly feasible target for halting the ongoing progression in T1D. However, more meticulous investigation is a prerequisite to identify the benefit-to-risk ratio, which seems more delicate in the case of children suffering from T1D. Additionally, the translation from preclinical to clinical studies has shown substantial variations, further rendering this approach a far-fetched idea.
4.4.1.3 Cytokine-Based Therapy
Cytokines are small, cell signaling molecules which orchestrate different processes such as T-cell growth (interleukins IL-2, IL-4, IL-7, IL-15, and IL-21), inflammatory responses by Th1/Type 1 cytokines (IL-1, TNF-α, TNF-β, IFN-α, and IFN-γ), and anti-inflammatory function by Th2/Type 2 cytokines (IL-4, IL-10, and TGF-β), respectively. Further, Th2 cytokines are involved in the expansion of Treg cells. The binary role of cytokines as mediators and regulators opens new prospects for T1D therapy. Some paradoxes exist the assumed inflammatory role of Th1 cytokines, since the administrations of IL-2 and IFN-γ are actually found to prevent T1D in animal models (Rabinovitch and Suarez-Pinzon 2007).
Interleukin 1 (IL-1)
IL-1, a pro-inflammatory cytokine, is a significant and overexpressed cytokine gene in peripheral blood mononuclear cells (PBMC) in patients with newly diagnosed T1D compared to healthy controls (Kaizer et al. 2007). In NOD mice, systemic knockout of IL-1 receptor gene was found to reduce diabetes mellitus rates by 30 % (Thomas et al. 2004). Anti-IL-1 (Anakinra) is a recombinant, non-glycosylated human version of anti-IL-1 receptor antagonist, clinically approved for the treatment of rheumatoid arthritis. In 2007, a clinical trial showed that blockade of IL-1 pathway through the use of Anakinra improved glycemia, β-cell secretory function, and reduced markers of systemic inflammation in T2D (Larsen et al. 2007). Recently, a study related to use of anti-IL-1 therapy in children with newly diagnosed T1D was conducted. Preliminary results showed that anti-IL-1 therapy is well tolerated in children. A similar level of HbA1c and mixed-meal tolerance test was observed in the treated and control groups, with additional low dose of insulin requirement in treatment group, 1 and 4 months post-diagnosis (Sumpter et al. 2011). The limitations of this study include non-randomized experiments, limited sample size, brief duration of therapy (28 days), independent controls groups (selected differently), and no precedent study to support the therapy duration. Nevertheless, the study suggests the need for randomized, placebo-controlled study over a longer duration in order to evaluate the true capacity of anti-IL-1 therapy to augment the insulin secretory capacity of T1D patients. Another study including anti-IL-1 mAb (Canakinumab) is currently in progress (ClinicalTrials.gov ID: NCT00947427).
Interleukin 2 (IL-2)
IL-2 is one of the cytokines exhibiting several functions such as T-cell growth, cytotoxic effects, and maintenance of Treg cells. Malfunctioning of IL-2 signaling in CD4+ T-cells of T1D subjects leads to lowered Foxp3 expression on the surface of Treg cells, lowering their tolerance effect (d’Hennezel et al. 2010). Bearing this theory in mind, IL-2 was tested preclinically and was found to be dose dependent. It was observed in NOD mice that low-dose administration of IL-2 resulted in increased induction of Treg cells with higher expression of Treg-related proteins such as Foxp3, CD25, CTLA-4, ICOS (inducible T-cell costimulator), and GITR (glucocorticoid-induced TNF receptor), and prevented T1D. Furthermore, IL-2 administration suppressed IFN-γ production (Grinberg-Bleyer et al. 2010). A cytolytic chimeric IL-2 and Fc fusion protein with specific binding to IL-2 receptor was found to suppress induced T1D in NOD mice (Zheng et al. 1999), while the Fc portion of the protein contributed to lowered immunogenicity and increased half-life (t1/2). According to Clinical Trials.gov, a study was recently completed using IL-2 (Aldesleukin) (study ID: NCT01353833) aimed at assessing its dose–effect relationship on T1D with results yet to be declared (http://clinicaltrials.gov/ct2/show/NCT01353833). A further study using a combination of IL-2 and rapamycin has shown limited success (Long et al. 2012). In contrast to the limited success with cytokines treatment, blockade of pro-inflammatory cytokines seems a more pertinent approach (Baumann et al. 2012).
Tumor Necrosis Factor-Alpha (TNF-α)
In addition to the aforementioned cytokines, TNF-α is additional pro-inflammatory cytokine which exhibits direct cytotoxic effects on β-cells. Anti-TNF therapy has gained wide recognition in the treatment of rheumatoid arthritis and Crohn’s disease, in particular. However, only recently it has been tested in various clinical trials evaluating their efficacy in T1D. In NOD mice, TNF-α or anti-TNF-α therapies have shown contrasting results in vitro and in vivo, with ablation of T1D in vivo as opposed to exacerbation of T1D or destruction of β-cells in vitro. This suggests that TNF-α might have a more potent immunoregulatory effects in vivo than a mediator effect (Jacob et al. 1990). In addition, the role of TNF-α in T1D pathogenesis remains enigmatic. One of the initial studies showed the age-dependent effect of TNF-α therapy in NOD mice. Administration of TNF-α (before 4 weeks of age) led to an earlier onset of T1D, whereas total blockade of TNF-α with mAb prevented T1D. The outcome clearly demonstrates the indispensable role of TNF-α in the thymic development of autoimmune repertoire, which could be due to its involvement in thymocyte activation and proliferation (Yang et al. 1994; Christen and Herrath 2002; Kodama et al. 2005; Koulmanda et al. 2012). In an attempt to study the feasibility and efficacy of anti-TNF-α therapy, a soluble recombinant TNF fusion protein (Etanercept) was used in a pilot scale study on 18 children with newly diagnosed T1D. The preliminary data showed prolonged effects on endogenous insulin production in these pediatric patients (Mastrandrea et al. 2009). Further, large-scale studies are underway to evaluate the true potential and associated side-effect profile with the use of this therapy.
Striking differences noticed with the use of cytokines reveal the need for careful contemplation based on (i) time of administration, (ii) route of administration, (iii) dose, and (iv) site of expression (local, i.e., organ-specific or systemic) (Bresson and Von Herrath 2007). A strategy such as localization of cytokine therapy directly to the site of islet inflammation is also something which needs attention, so as to avoid nonspecific and undesirable side effects in vivo. In summary, these data mark the complexity of using cytokine alone as immune intervention. Finally, there is an enormous potential that the anti-cytokine treatment, along with other therapies, may prevent or delay diabetes development in individuals at high risk of the disease.
4.4.2 Antigen-Based Intervention Strategies
Antigen-based therapy is a developing field and currently relishes the most attention. The rationale following its investigation is antigen specificity which adds to its safety profile, something not found with immunosuppressive agents or nonantigen-specific immunomodulation approaches (Ludvigsson 2009). However, these therapies suffer drawbacks such as unpredictable outcome of immunization that depends on numerous factors such as antigen dose, frequency, and route of administration (Peakman and von Herrath 2010).
Treatment using autoantigen delivery is a well-known concept and dates back to the twentieth century when it was used to treat hypersensitive allergies toward innocuous foreign antigens (Krishna and Huissoon 2011). The specific induction of Tregs in response to administration of an exogenous antigen has been termed inverse or negative vaccination (Mercer and Unutmaz 2009; Hinke 2011). By poorly understood mechanisms, naive T-cell activation matures either into Teff or CD4+CD25+ T-cell/Treg cells. As we know from our previous discussion, Teff cells are responsible for induction of an immune response. Tregs can be further classified as (i) natural, i.e, thymus derived (nTreg) or, (ii) adaptive, i.e, induced within the periphery (iTreg). These Tregs are essential for the maintenance of immune tolerance. In some cases, the Foxp3, a marker of Treg cells, shows increased expression on these cells. Unfortunately, these Foxp3 are murine-specific Treg markers, because their utility in humans is restricted as they are additionally expressed on recently activated Teff cells. The fate of T-cells depends on antigenic signal strength and prevalent cytokines. The underlying mechanism behind this therapy is the tendency of autoantigens to either delete the effector/pathogenic (Th1 and CTLs) and autoreactive (CD4+ and CD8+) T-cells or their ability to expand protective Treg specific to the autoantigens (Bresson and von Herrath 2009). Several Treg-associated mechanisms have been implicated in the development of immune tolerance by antigen-specific therapy. First, bystander suppression which refers to specific antigen induced generation of Treg cells that nonspecifically alter the function of APC and hence suppress inflammation in the target organs by suppressing the Teff cells induced by other antigens (Anderton et al. 1999; Millington et al. 2004). Second, infectious tolerance which means Treg cell clone has a natural ability to induce Tregs of different antigen specificities (Li 2011; Harrison 2012).
Several ways are available to achieve antigen-specific therapy of T1D in animal models: (i) use of full length islet proteins (such as GAD65), (ii) altered peptide ligands derived from islet antigens (such as heat shock protein 6), and/or (iii) with DNA vaccines (InsB9-23) (Slobodan et al. 2011). Various autoantigens associated with T1D like hsp60/Diapep277, insulin (Ins), and GAD65 (Diamyd®) have been tested in clinical trials with varied levels of success. The next section includes details of the updated knowledge of different antigen-specific therapies.
4.4.2.1 Insulin Vaccine
Insulin and proinsulin epitopes have been identified as playing a vital role in the early stages of T1D autoimmune processes both in humans and NOD mice models (Zhang et al. 1991; Kent et al. 2005; Nakayama et al. 2005). Insulin autoantibodies (IAA) precede the clinical presentation of T1D especially in children and are often used as biomarkers in disease prediction. Insulin therapy has shown a twofold benefit: (i) it restores the insulin-specific immune tolerance and, (ii) it halts ongoing β-cell destruction with the supply of active, exogenous insulin, and thus (iii) it reduces the insulin secreting stress on the pancreas. Based on the large body of evidence which shows insulin’s role in T1D, inverse vaccination with insulin and proinsulin either as a peptide or DNA vaccine seems like an ideal and relatively facile approach for prevention or intervention in T1D (Hilsted et al. 1995; Skyler 2008). Table 4.3 includes various trials that are complete or in ongoing stages using insulin as a drug candidate.
Table 4.3
Outline of prevention and intervention trials using insulin/proinsulin as a target antigen
Study/clinical trial | Therapy stage | Antigen | Dose/follow-up period/route of administration | Study group/age group | Study outcome | Lesson learnt | References |
|---|---|---|---|---|---|---|---|
Pre-POINT | Primary prevention | Oral/intranasal insulin | • Insulin daily for 1st 10 days followed by twice dose every week • 2.5–67.5 mg (Oral) • 0.28–7.5 mg (Intranasal) Mean follow-up of ~3–18 months | 1st degree T1D relatives with >50 % risk of T1D Age: 1.5–7 years | Recruiting | • Dose escalating study Intend to find optimal intervention timing and dose | Rewers and Gottlieb (2009) |
DPT-1 (1st arm) | Secondary prevention
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|