Historical background
Turner syndrome (TS) is defined as a disorder caused by complete or partial absence of the second sex chromosome, with or without cell line mosaicism. This further needs to be associated with one or more characteristic physical features in a phenotypic female, such as short stature or primary ovarian insufficiency. The eponym comes from a study published in 1938 by Henry Turner, who described seven women with short stature, sexual infantilism, neck webbing, a low posterior hairline, and an increased carrying angle or cubitus valgus ( Fig. 17.1 ). Several years earlier (in 1930), the clinical geneticist Otto Ullrich had independently described an 8-year-old girl with short stature, lymphedema of the hands and feet, neck webbing, a high arched palate, low-set external ears, and some other characteristics that are now recognized features of TS. Ullrich subsequently recognized that his patient and those of Turner probably had the same condition. He further called attention to the work of Bonnevie, who described neck distention and malformations of the ears, face, and limb buds in mice, secondary to dissection of subcutaneous fetal tissues by fluid. Ullrich suggested that fetal lymphatic obstruction may cause neck webbing and other features of TS and proposed the eponym Bonnevie-Ullrich to describe this constellation of anomalies. Ullrich’s contributions gave rise to the eponym Ullrich-Turner syndrome more commonly used in Europe.
Endocrine and pathology studies performed already in the 1940s revealed the presence of primary ovarian failure in TS women, further associated with elevated gonadotropins, reduced estrogen concentrations, and “streak” ovaries, consisting of connective tissue depleted of germ cells. These early studies also described an increased incidence of hypertension and aortic disease in young TS individuals. The first link between TS and a sex chromosome anomaly was provided in 1954 by Polani and colleagues, who reported on three women with TS and coarctation of the aorta, while also being sex chromatin negative. Soon thereafter, advances in cytogenetic identification of specific chromosomes revealed that TS was associated with the presence of a single X chromosome (X monosomy). These observations signified a major shift in our appreciation of the role of the human sex chromosomes in sex determination, as reviewed by Opitz and Pallister. They also described the significant heterogeneity of patients grouped under the umbrella of gonadal dysgenesis. Dysgenesis and agenesis represent inaccurate descriptions for TS individuals’ ovarian histopathology, because fetal ovarian development appears normal in TS, and the ovarian tissue degeneration occurs mainly around the time of birth. Although eponyms have their disadvantages, the designation Turner (or Ullrich-Turner) syndrome is also more specific than gonadal dysgenesis .
Genetics
Chromosomal Origins
In 1959 TS was linked to monosohmy X in a 14-year-old girl by using a karyotype of colchicine-arrested metaphase bone marrow cells. In a subsequent study of 307 individuals with gonadal dysgenesis and sex chromosome anomalies, the short arm of the X chromosome was identified as the critical region responsible for TS .
Today, 6 decades after the first karyotyping of TS, the molecular mechanisms that lead to monosomy X and TS are still not completely understood. Based on other aneuploidies, especially the trisomies, such as Down syndrome, the most popular hypothesis for the molecular basis of TS is an error in meiosis. A maternal X is present in 70% to 80% of individuals with TS, suggesting a male predisposition to meiotic errors. Fluorescent in situ hybridization (FISH) studies in sperm support a male predisposition to meiotic errors in the sex chromosomes as autosomal disomy occurs in 0.1% and sex chromosome disomy occurs in 0.27%. If indeed TS results from an error in meiosis, unlike for trisomy investigations, the stage of meiosis that contributes to monosomy X cannot be determined. For example, in Down syndrome, nonsegregation is known to occur in meiosis II, when two chromosome 21s are homologous.
Besides an error in meiosis, the other possibility explaining monosomy X is a postzygotic nonsegregation in mitosis. Postzygotic nonsegregation is an attractive hypothesis considering that approximately half of individuals with TS are mosaic. However, there are no empirical data in humans to support this hypothesis. However, there is evidence in the mouse model that the Y chromosome is predisposed to nondisjunction during early cleavage division, and more research in humans should provide further answers.
In addition to nonsegregation in meiosis and mitosis, there are a number of structural anomalies of the X chromosome, including isochromosome X [46,X,i(Xq)] and partial deletion of Xp and Xq. The X isochromosome is the most common isochromosome in humans, and it is likely caused by chromosome breakage and recombination in the proximal Xp, resulting in an isodicentric X i(Xq) chromosome.
Epidemiology
TS is a common finding in newborn females with a birth incidence of 1 in 2500. However, TS is diagnosed more frequently prenatally. Using the Danish Cytogenetic Central Register, Gravholt et al. compared the prenatal prevalence of TS to its postnatal prevalence. A postnatal prevalence of 32/100,000 was found compared with 176/100,000 in diagnoses by amniocentesis and 392/100,000 by chorionic villus sampling (CVS). Wharton and Hook, in their study of the New York state registry, also found a higher prevalence in prenatal diagnoses: 22.2/100,000 and 85/100,000 in postnatal age and prenatally by amniocentesis, respectively.
Turner Karyotypes
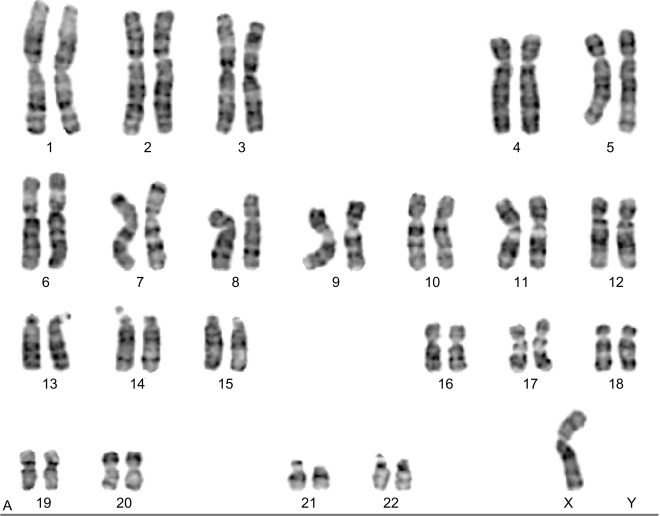
Most karyotype determinations are made from mitotic peripheral lymphocytes that have been arrested in metaphase or prometaphase. The limitation of the peripheral blood karyotype is that only blood is tested and no other tissues, thus mosaicism cannot be tested reliably. A second tissue, such as skin fibroblasts, or buccal mucosa cells, or bladder epithelial cells, should be evaluated when a peripheral blood karyotype is normal and TS is still suspected. A large Danish study has set the fractions of various peripheral blood karyotypes and found that in 781 individuals with TS: 45% were found to have 45,X; 11% had a karyotype that included an isochromosome, such as 46X,i(Xq) or 45,X/46X,i(Xq); and the remaining 44% were mostly mosaic, such as 45,X/46,XX. Supporting further a high rate of mosaicism in TS, a review of 532 live born girls and women with TS found that approximately half were mosaic.
From a clinical and genetic counseling perspective, it is important to consider why and when a karyotype is obtained, as the timing of karyotype analysis often correlates with the phenotype severity in TS. Many prenatal karyotypes diagnosing TS have been done for advanced maternal age and compared with postnatal diagnoses, these individuals have decreased cardiac anomalies and decreased incidence of phenotypic features, such as neck webbing and low hairline, while characterized by a higher incidence of mosaicism. For the purpose of genetic counseling of 45,X/46,XY mosaics, it is important to highlight that in a study of 92 cases of prenatally diagnosed 45,X/46,XY mosaic cases, 95% had normal male genitalia.
A significant finding on a karyotype is the presence of Y chromosome material, which has implications in clinical management. A Y chromosome detected by karyotype is a risk for gonadoblastoma. In a study of 114 females with TS, 14 (12.2%) had Y material found by polymerase chain reaction (PCR), of which seven had detectable Y chromosome material on karyotype. Interestingly, of the 10 individuals in this study who underwent ovariectomy, only one (10%) had a gonadoblastoma. In another study of 171 individuals with TS, 14 (8%) were found to have Y chromosome material by Y chromosome repeat markers, and four of the 12 individuals who had a gonadectomy had gonadoblastoma detected. In a more recent study of 130 individuals with TS, three individuals were found to have Y chromosome material by karyotype and six by reverse transcription-PCR. Of these nine individuals with Y chromosome material, all underwent gonadectomy and one was found to have a gonadoblastoma. Although the aforementioned studies are heterogeneous in their methods, the risk of gonadoblastoma based on the best evidence available is roughly 10% if Y chromosome material is present. Current recommendation is for gonadectomy to be performed in TS individuals with Y chromosome material found on a standard karyotype analysis ( Fig. 17.2 ).
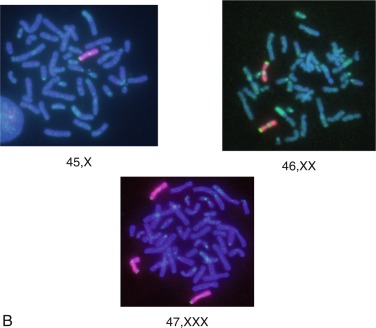
X Chromosome Genes and Turner Syndrome
Genotype-Phenotype Associations
Over 160 million years ago, the X and Y chromosomes were homologous, until the Y chromosome acquired the sex determining gene SRY and slowly lost many of its genes because of isolation from recombination by inversion. A dosage compensation system evolved to equalize expression of the X chromosome genes on the 46,XX female and 46,XY male. The compensation mechanism is X chromosome inactivation (XCI), which is achieved by the ribonucleic acid (RNA) coding gene XIST expressed on the inactive X chromosome; XIST then coats the inactive X chromosome making it inactive. However, 20% to 30% of X-linked genes escape X inactivation and are expressed on the inactive X chromosome. Pseudoautosomal region 1 (PAR1) located at the distal tip of the short arm of the X chromosome (Xp) and the short arm of chromosome Y (Yp) contains 24 genes that escape this X inactivation. The pseudoautosomal region (PAR) is divided into two regions, PAR1 at the terminus of the short arm of X and Y, and PAR2 on the long arm. The PARs pair and recombine during meiosis. There are four genes on PAR2 on both the X and Y chromosomes, and these genes, interestingly, do not escape X inactivation on the X chromosome. There are further 17 X-Y ancestral homologs, of which also 14 escape X inactivation. However, the remaining majority of X-linked genes that escape XCI do not have homologs on the Y chromosome. Genes that escape XCI are haploinsufficient in TS and have historically been candidates for genotype-phenotype investigations. However, few X genes have been correlated with the TS phenotype. The short stature homeobox containing gene on the X chromosome ( SHOX) is the most cited example ( Fig. 17.3 ).
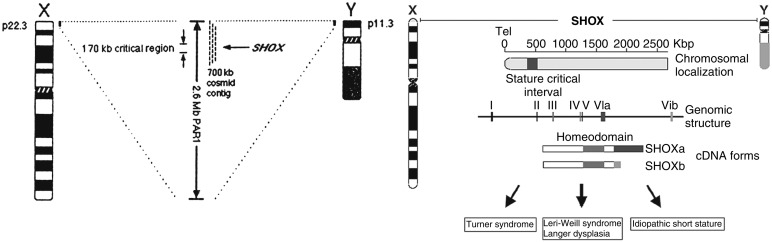
SHOX is located at Xp22 on the pseudoautosomal region of the X chromosome. Both males and females have two copies of SHOX. SHOX , a transcription factor, has been implicated in short stature and the skeletal anomalies associated with TS. In a study of over 1600 prepubertal non-TS children with short stature, deletions or pathogenic variants in SHOX were found in approximately 4% of individuals. SHOX heterozygous variants, most commonly a deletion, may cause isolated short stature or other anomalies, such as mesomelia or the Madelung wrist deformity, observed in Leri-Weill dyschondrosteosis. The concept of haploinsufficiency of a gene, such as SHOX located in the PAR, is an attractive explanation for part of the TS phenotype. However, there is currently little evidence of any other monogenic variants on the PARs associated with the TS phenotype.
In addition to short stature, infertility and primary ovarian insufficiency are phenotypes for which a genetic etiology has been sought. Mapping partial X chromosome deletions in women, with and without TS, has provided loci associated with primary ovarian insufficiency (POI). Starting with the short arm of the X chromosome (Xp), TS phenotypes including POI have been mapped to Xp11.2-Xp22.1. In non-TS women, there is one gene located in this locus that has been associated with POI. Bone morphogenetic protein 15 ( BMP15 ) , located on Xp11.2, is an oocyte growth factor that stimulates folliculogenesis and granulosa cell (GC) growth, and has been implicated in POI. Di Pasquale et al. presented two sisters with a heterozygous missense variant in BMP15 (p.Y235C) and functional studies suggested a dominant negative mechanism. In a more recent study of 300 women with POI, six variants were found in BMP15 with three of the variants leading to marked decrease in protein production. Interestingly, there is a report of a small 554kb duplication involving the genes BMP15 and SHROOM4 in a woman with TS and spontaneous menarche, adding more evidence to BMP15’s association with POI.
On the long arm of the X chromosome (Xq), a number of X chromosome-autosome translocations in non-TS women have identified two additional loci or critical regions associated with POI: Xq13-Xq21 (critical region I) and Xq23-Xq27 (critical region II). In these critical regions, a small number of women without TS have shown associations with genetic variants and POI. In an X-autosome translocation [t(X;11)(q24;q13)] found in both a mother and daughter with POI, disruption in the gene progesterone receptor membrane component-1 (PGRMC1) was found, based on reduced gene expression levels. PGRMC1 mediates antiapoptotic action of progesterone and several cytochrome P450 reactions in ovarian cells. Mansouri et al. screened 67 females with POI and found one person with a p.H165R variant that was shown to interfere with cytochrome P450 7A1 (CYP7A1) binding to PGRMC1. In a large database of over 43,000 presumed healthy individuals, this particular variant was found in 0.2%. Located in Xq21, a report of five daughters with POI to a consanguineous couple identified a homozygous variant in premature ovarian failure protein 1B (POF1B ) . This variant was also shown to affect nonmuscle myosin binding. One prior report in a woman with POI demonstrated a translocation involving chromosome 1 and the X chromosome [t(X;1)(q21;p34)] with a breakpoint in the third intron of POF1B . POF1B is of interest as it escapes X inactivation and is located in POI1. Also located in critical region I, dachshund homolog 2 ( DACH2) was first identified by fine-mapping the breakpoint of an autosome-X chromosome translocation in a patient with POI. Two missense variants in DACH2 have been associated with women with POI. However, there is no functional studies’ evidence to support DACH2’s role in POI.
Although not in the two critical loci noted earlier, two missense variants in androgen receptor gene (AR ) on Xq12 have been associated with POI in Indian women with POI, but the study did not evaluate these variants with functional studies. The androgen receptor is of potential interest as deficiency of AR in the mouse model results in POI.
A gene on the X chromosome worthy of special mention is fragile X mental retardation (FMR1) on Xq27.3. A premutation, 50–200 CGG repeats in the 5′ untranslated CGG repeat, of FMR1 on Xq27.3 leads to methylation-coupled silencing of FMR1 and absence of the fragile X mental retardation protein (FMRP), causing the classical Fragile X syndrome. Fragile X syndrome is a common X-linked cause of cognitive impairment in males when the CGG repeat is greater than 200. Although haploinsufficiency of FMR1 is unlikely to cause POI in TS, a premutation is a common cause of POI, explaining 5% of sporadic POI cases and 10% to 15% of familial cases. The mechanism of POI in FMR1 premutation is thought to be an increase in messenger RNA (mRNA).
Short stature and POI are the most studied phenotypes in TS, and cognitive impairment will only be discussed briefly. The gene XIST has been associated with cognitive impairment in TS. Cognitive impairment is infrequently associated with TS and has mostly been observed in individuals with mosaic (short) ring chromosome [45,X/46,X,r(X)]. Cognitive impairment was first described in nine of 21 individuals with such a ring chromosome. The initial hypothesis for cognitive impairment was thought to be absence of the XIST gene on the ring chromosome, resulting in the failure of the X-inactivation of the ring chromosome. However, Leppig noted that in nine individuals with an r(X), eight had XIST genes present, questioning the original “absence of XIST ” hypothesis.
It must be emphasized that genotype-phenotype relationships in TS remain largely unknown. New approaches, such as methylation- and RNA expression studies are beginning to provide new answers. In a lymphocyte methylation and RNA expression study comparing TS women and 46,XX and 46,XY controls, women with TS were found to have genome wide hypomethylation altered expression in genes on both the autosomes and sex chromosomes compared with controls. Other studies have identified X chromosome microRNA that escapes X-inactivation and may play an important role in disease differences between males and females.
X Chromosome Genomic Imprinting
Imprinting studies for TS have shown mixed results. Skuse et al. proposed that individuals with 45,X who inherited a paternal X chromosome had a higher social cognition, meaning a significant difference in verbal and higher-order executive function skills. This study was based on 80 individuals with TS, of whom 55 had inherited a maternal X. In a smaller study of 39 participants with TS, Bishop et al. found that immediate and delayed recall was in the normal range for girls with TS. A study that looked at brain morphology in 40 girls with 45,X (23 girls with a maternal X and 17 with a paternal X) found differences in brain morphology based on X chromosome origin: those who inherited a paternal X had a thicker cortex in the temporal regions, and those who inherited a maternal X had enlargement of gray matter in superior frontal regions. Lepage et al. also noted that girls with a paternal X had a lower full-scale IQ than those that had inherited a maternal X. In contrast to these studies, a study of 50 girls with TS showed no association between parental X and IQ scores, as determined by the Wechsler Intelligence Scale for Children. Finally, for the most recognized phenotype of short stature, there is no evidence of X chromosome imprinting based on a small study of 25 individuals with TS.
Diagnostic Tests
There are multiple testing options available for TS diagnosis, both prenatally and postnatally. As noted earlier, the first diagnosis was made in 1959 by karyotype testing and this method continues to be the standard of care. A karyotype can be made from any metaphase or prometaphase cell that is actively dividing, including peripheral blood lymphocytes, amniocytes, and fibroblasts. Chromosomal microarray has the same level of accuracy as a karyotype, but has the advantage of more precise chromosomal breakpoints in the case of partial deletions and translocations. Although not a first-line test for TS, next generation sequencing can accurately diagnose TS and has the advantage of being able to simultaneously diagnose single gene variant conditions such as Noonan syndrome. FISH may be used to diagnose TS and has the advantage of being faster than karyotype testing, and can be performed on nondividing cells, such as those derived from buccal swabs.
Indications for Karyotype Testing
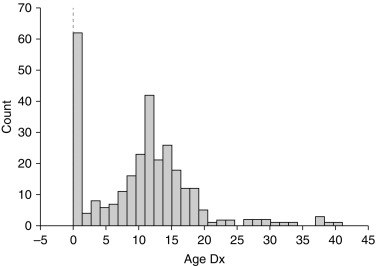
There is a distinct biphasic pattern in the diagnosis of Turner syndrome with a substantial proportion being diagnosed around the time of birth, and another large group being diagnosed around 12 years of age ( Fig. 17.4 ). When there is a clinical suspicion of TS, the American College of Medical Genetics recommends a 20-cell karyotype that should be able to detect mosaicism greater than 11%. For a prenatal 45,X karyotype diagnosis, TS should be confirmed with a postnatal karyotype .
Differential Diagnosis
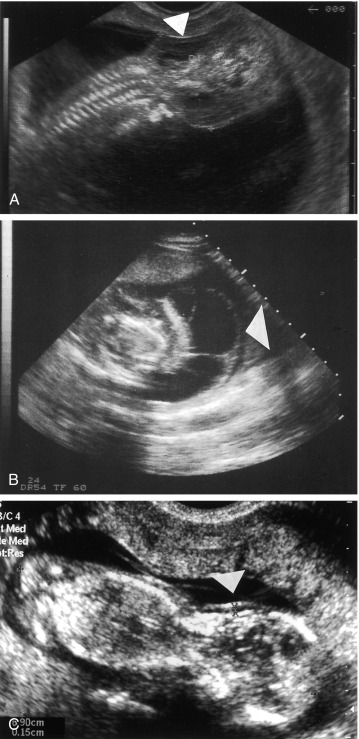
When TS is suspected prenatally or postnatally, other genetic etiologies should be considered. In the prenatal period, TS may be diagnosed with noninvasive prenatal testing (NIPT) or CVS. Confined placental mosaicism, when the placenta is 45,X and the fetus is euploid, is in the differential diagnosis and should be resolved with amniocentesis. In addition to NIPT and CVS, prenatal ultrasound findings may suggest TS and other syndromes ( Fig. 17.5 ). The following prenatal ultrasound findings may be found in other genetic syndromes: hygroma colli, fetal hydrops, coarctation of the aorta, and increased nuchal translucency. In first trimester ultrasound of 185 singleton pregnancies with cystic hygroma colli, the most frequent chromosomal abnormality was TS ( n = 49; 26.5%), followed by trisomy 21 ( n = 32; 17.3%), and trisomy 18 ( n = 27; 14.6%). In this same study, 15 euploid fetuses were tested for Noonan syndrome and six (40%) were found to have it. In 65 cases of hydrops fetalis, six (9.2%) were diagnosed with TS, three (4.6%) with trisomy 21, and two cases with Noonan syndrome (3%).
Postnatally, TS may be suspected in childhood because of short stature, or during the pubertal age because of (primary) amenorrhea. Short stature affects 3% of children and there are numerous genetic etiologies. Common etiologies on the differential diagnosis include Noonan syndrome and haploinsufficiency for the SHOX gene. Although Noonan syndrome has been labeled “male TS” by some in the past, the facial, cardiac, and development features are quite different between these two syndromes.
Prenatal Diagnosis
NIPT has changed the timing and accuracy of genetic prenatal screening, especially for aneuploidy syndromes, such as TS. NIPT is based on small fragments of circulating cell-free fetal deoxyribonucleic acid (ccfDNA) that originate from placental cells that are continuously released, and can be detected as early as 5 weeks of gestation. Approximately 10% to 15% of cell free DNA in the maternal circulation is from the placenta, when measured between 10 and 20 weeks of gestation, and the half-life of ccfDNA is less than a day. There are a number of methods using NIPT to screen for TS prenatally, and most techniques count the number of copies of sequenced DNA fragments using next generation sequencing techniques.
In a Cochrane database systematic review, a pooled analysis of over 7000 high-risk pregnancies involving 45,X, in 12 studies using massive parallel shotgun sequencing, showed a clinical sensitivity of 91.7% (95% confidence interval [CI], 78.3–97.1) and a clinical specificity of 99.6% (95% CI, 98.9–99.8). The Cochrane database study found similar results for roughly 1000 high-risk individuals in four studies, using targeted massive parallel sequencing, which only evaluates select loci of the genome. Even with high sensitivity and specificity, the positive predictive values for NIPT for TS may be as low as 23% to 26%, which strongly reinforces the need for confirmatory testing. The American College of Obstetricians and Gynecologists recommends that all women with a positive cell free DNA result have a diagnostic procedure before clinical measures, such as pregnancy termination is taken.
The diagnosis of monosomy X is possible in embryos formed after in vitro fertilization (IVF) and is termed preimplantation genetic diagnosis ( PGD ) or preimplantation genetic screening . A biopsy of a blastomere at the cleavage stage or a trophectoderm biopsy at the blastocyst stage allows for an aneuploidy assessment, using techniques including FISH, chromosomal microarray, and next generation sequencing. PGD analysis of IVF embryos is motivated by the belief that selection of euploid embryos will improve IVF outcome. However, to date, there is no evidence that PGD improves the live birth rate after IVF. More research is needed in PGD applications to aneuploidy, especially with respect to diagnosing TS.
Clinical characteristics and comorbidities
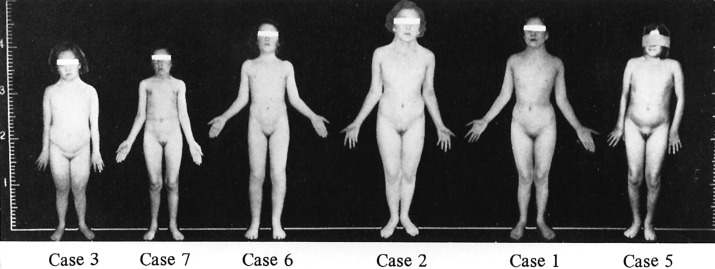
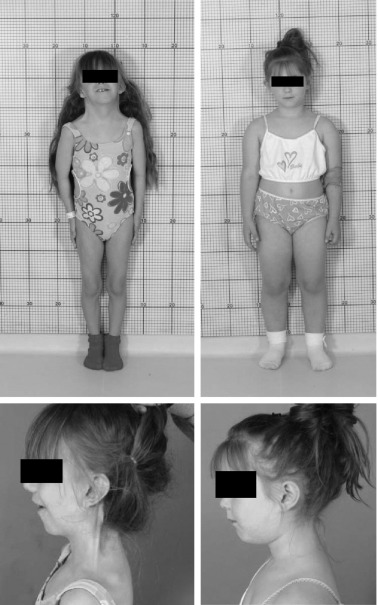
Individuals with TS may present with a variety of phenotypic features: some patients have few and others are characterized by a multiplicity of findings. The typical physical characteristics can vary significantly between patients with similar karyotype ( Fig. 17.6 ), and some of the clinical signs may be subtle. Table 17.1 summarizes the clinical features most commonly encountered in girls and women with TS.
| Physical Diagnosis | Percentage Affected | |
|---|---|---|
| Skeletal | Short stature | 100 |
| Short neck | 40 | |
| ↑ Upper:lower body ratio | 97 | |
| Cubitus valgus | 47 | |
| Short metacarpal | 37 | |
| Scoliosis | 12.5 | |
| Madelung deformity | 7.5 | |
| Micrognathia/high palate | 60 | |
| Lymphatic obstruction | Neck webbing | 25 |
| Low posterior hairline | 42 | |
| Edema of hands/feet | 22 | |
| Nail dysplasia | 13 | |
| Other | Strabismus | 18 |
| Ptosis | 11 | |
| Multiple nevi | 26 | |
| Screening Evaluation | ||
| Cardiovascular anomaly | All | 44 |
| Bicuspid aortic valve | 30 | |
| Aortic coarctation | 12 | |
| Dilated aorta | 11 | |
| Other a | 12 | |
| Renal anomaly | All | 18 |
| Horseshoe kidney | 11 | |
| Duplicated collecting ducts | 4 | |
| Unilateral agenesis | 3 | |
| Liver disorder | All | 36 |
| Abnormal liver function tests (LFTs) | 27 | |
| Fatty infiltration | 19 | |
| Hypertension | All | 34 |
| Prehypertension | 14 | |
| Overt hypertension | 20 | |
| Autoimmunity | All | 51 |
| Hashimoto thyroiditis | 51 | |
| Graves disease | 1 | |
| Type 1 diabetes | 0 | |
| Celiac | 5 | |
| Inflammatory bowel | 3 | |
a Under the cardiovascular listing, the “Other” category included partial anomalous pulmonary veins, aberrant right subclavian artery, and atrial septal defects. Blood pressure was measured on 24-hour ambulatory monitors. All patients also had renal and hepatic ultrasound studies. Abnormal liver function was defined as greater than 10% elevation of aminotransferase(s). Hashimoto was defined by history of clinical hypothyroidism or elevation of circulating thyroid antibodies.
Lymphedema
Lymphedema is relatively common in TS and begins in utero. Fig. 17.7 demonstrates the generalized edema that can be observed in many 45,X fetuses. It is the consequence of malformations of the lymphatic vessels, resulting in lymphatic hypoplasia and obstruction. A cystic hygroma is a fluid-filled sac that often is located in the head/neck area, but may occur in other parts of the body, and results from abnormal communication between the jugular and central lymphatics draining into the heart. Peripheral lymphatic hypoplasia or aplasia has also been demonstrated. Using lymphangiography hypoplasia, and even aplasia, can also be observed in the peripheral lymphatic vessels of the extremities.

Clinical features resulting from nuchal lymphatic obstruction include webbing of neck, also called pterygium colli , and is observed in up to 25% of TS patients. Additional features that appear to result from this tenting and stretching of facial/scalp and neck skin is thought to result in low-set, rotated external ears, ptosis and downsloping of the eyelids, and the development of a low posterior hairline, with upward growing hairs (see Fig. 17.6 ). The occurrence of thick hair growth, including thick eyelashes and eyebrows, may also be, at least in part, a consequence of the distended cutaneous structures during fetal life. Swelling of the dorsum of the hands and feet at birth indicates the presence of a nonpitting type of peripheral edema. The lymphedema usually improves over the first 1 to 2 years of postnatal life. It may even resolve, but in many patients it persists to some degree. Some TS individuals demonstrate intermittent worsening of their peripheral edema, and this appears to be associated with puberty or the introduction of growth hormone (GH) or sex steroid therapy (both somewhat salt-retaining).
Growth failure, Short Stature, and Skeletal Anomalies
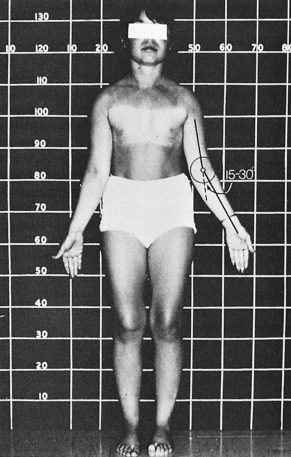
Short stature is the most common physical abnormality seen in TS. Poor statural growth affects essentially every patient to some degree. Their growth failure is disproportionate and they have a relatively large trunk with broad shoulders and pelvis; together with the reduced height this renders them with a “stocky” appearance. They often have a short neck because of hypoplasia of one or several cervical vertebrae. Long bone growth is preferentially impaired: patients do have relatively large hands and feet relative to their height, but the relatively short lower extremities lead to an increased upper-to-lower segment ratio. Selected bones can be more affected than others: an increased carrying angle at the elbow joint, also called cubitus valgus , is observed in nearly half of TS individuals. This carrying angle is the angle of intersection of the long axis of the upper arm with the long axis of the fully extended and supinated forearm. In adult normal females, this elbow angle is approximately 12 degrees (twice the size as seen in males). This angle is between 15 and 30 degrees ( Fig. 17.8 ) in many girls and women with TS because of abnormal development of the head of the trochlear bone.
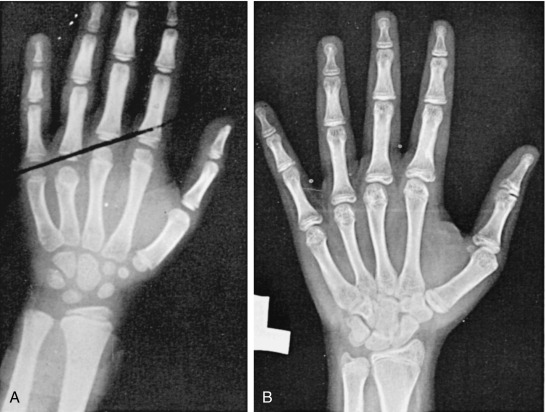
The finding of a shortened fourth metacarpal, as shown on a bone age radiograph ( Fig. 17.9 A ) is also quite common. On physical examination, it manifests as absence or less prominence of the fourth knuckle when making a fist. Sometimes the fifth knuckle is also affected. Short fourth metatarsals can be found is some patients. The Madelung deformity is less common and is caused by a curvature of the distal radius in addition to a dorsal subluxation of the distal ulna ( Fig. 17.10 ). This anomaly also occurs as part of Leri-Weill dyschondrosteosis, and in both conditions is an indicator of a deficiency of SHOX. The previously described cubitus valgus and short metacarpals are also the result of SHOX deficiency.

At least 10%, and possibly as many as 20% of TS girls between 5 and 20 years of age develop scoliosis, defined as a lateral curve greater than 10 degrees. Kyphosis may develop in up to 40% of patients. Both findings likely are the result of vertebral development anomalies. Abnormalities of the development of facial bones and skull base result in a micrognathic and retrognathic mandible and/or high arched palate in 60% of patients.
Osteoporosis and fractures are more common in women with TS. Hand and wrist radiographs may reveal mild osteopenia ( Fig. 17.9 B), a coarse trabecular bone pattern, and ballooning of the tips of the terminal phalanges. This osteoporotic appearance is observed during childhood and adolescence, suggesting it may be related to a developmental role of SHOX inherent to TS rather than to primary estrogen deficiency, which may still be a contributory factor. The fact that cortical bone mineralization is selectively reduced in TS, independent of estrogen exposure, is consistent with this. Fractures of the long bones not associated with significant trauma are more frequently observed among girls with TS. It should be noted, however, that interpretation of studies of bone mineralization in young individuals with TS is difficult because areal instead of volumetric bone mineral density is measured. Nevertheless, prepubertal TS girls have decreased markers of bone formation because of a state of low-bone turnover and decreased bone deposition.
Poor linear growth in childhood and adolescence leads to adult TS women ending up about 20 cm shorter than their non-Turner female peers. The short stature in TS can be attributed to the deleterious effect of SHOX haploinsufficiency.
One of the first large-scale assessments of the growth failure present in TS was reported by Ranke et al. in 1983. Fig. 17.11 shows cross-sectional height and height velocity data from 150 TS children who did not receive growth-promoting therapy. Davenport and coworkers further elaborated on the different stages of impaired growth throughout a TS patient’s life. Prenatally, there is a mild degree of growth restriction resulting in a length at birth that is about 1 standard deviation (SD) below the mean. During infancy and up to 3 years of age, a further mild growth deceleration can be seen. Continued subnormal growth is observed throughout childhood, so that between ages 3 and 13 years, TS girls develop more deflection from both their target percentile and the normal height curves. There is failure to experience a normal pubertal growth spurt, and slow growth may be prolonged for several more years, potentially into the early 20s.
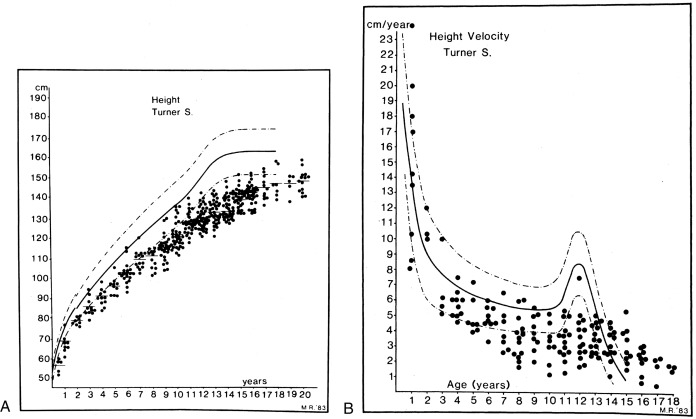
Height at diagnosis, ultimate height achieved, and midparental height are positively correlated. The adult height deficit is approximately 20 cm and this is irrespective of patients’ ethnic background. TS-specific growth curves have been developed by Lyon and associates, based on the data from Ranke and colleagues, as well as other European centers. These curves provide mean height and SD values for age and indicate a mean adult height of 143.1 cm (about 56.5 inches). A strong correlation between the initial height on these Turner curves and the adult height was also observed by Lyon and colleagues, independently of bone age at the time of the first height. This essentially allows for the adult height prediction of any girl with TS based on her height at an earlier age.
Ovarian Insufficiency
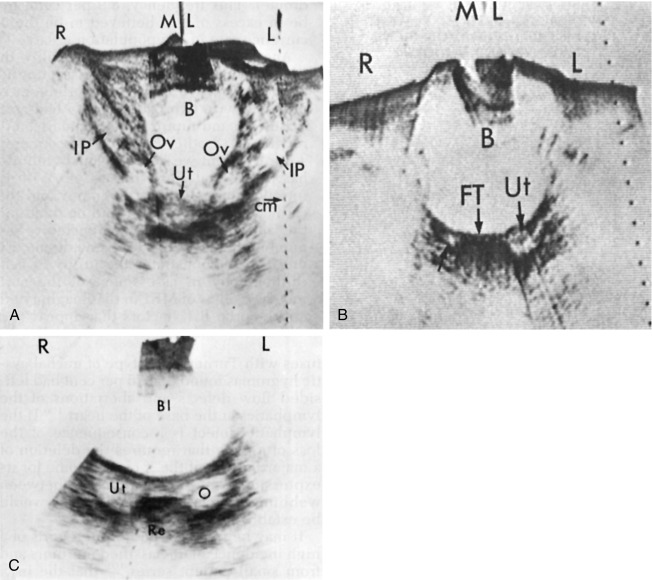
Initial reports on the presence of gonadal pathology described TS individuals as having streak ovaries. These small fibrous gonadal structures have few if any oocytes present. It appeared as if the ovaries had failed to develop normally. Subsequent studies revealed that early ovarian development actually occurs in a normal manner, with adequate numbers of oocytes and primordial follicles present in 14- to 16-week-old Turner fetuses. However, later in development, Turner ovaries become relatively depleted of oocytes and have fewer developing follicles. This suggests a process of accelerated oocyte demise and follicular atresia. Despite this, some TS girls have follicles in various stages of development, even during the second decade of life. Fig. 17.12 shows pelvic ultrasound studies comparing the pelvic structures of TS individuals with those of a normal pubertal females. The cause for the high rate of oocyte attrition in most girls with TS is unknown. One possible explanation is that premature oocyte depletion is caused by abnormal meiosis because of the aneuploidal status. Another possible explanation is that normal oocyte generation and survival require diploid expression of certain genes on the X chromosome. The chromosomal location of putative genes important for fertility has yet to be defined. This is because POI/gonadal failure is frequently observed in Xp deletions with an intact Xq complement, but also in some cases of Xq deletion with normal Xp complement. In addition, it has been reported that terminal Xq deletions can be associated with POI in women who have few, if any features of TS (see earlier section on Genetics, for more detailed description).
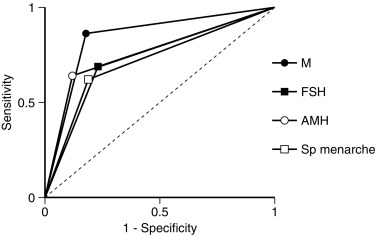
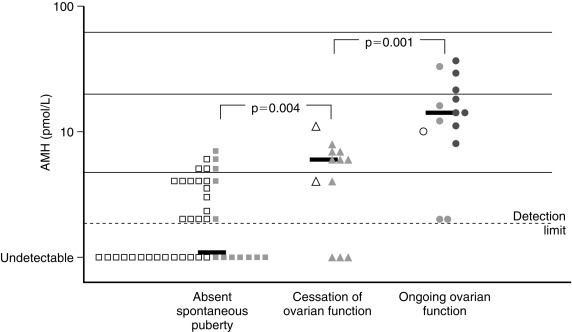
Because there is a broad range of gonadal dysfunction in TS individuals, it is hard to predict which patient will spontaneously enter puberty, and who will not, or to determine which women could become pregnant. Based on information from ovarian biopsies the strongest predictor for the presence of ovarian follicles is a karyotype with mosaicism for 45,X and 46,XX cell lines. TS arising from 45,X monosomy or structural defects of one X chromosome are much less likely to be associated with the presence of follicles in the ovaries. Additional positive predictors of follicle presence include both normal follicle-stimulating (FSH) and anti-Mullerian hormone (AMH) concentrations—see also Figs. 17.13 and 17.14 . Although less robust than the blood karyotype, the occurrence of spontaneous puberty is also a positive predictor. Spontaneous puberty was observed in approximately 15% of girls with 45,X monosomy and in 30% of girls with a second cell line with more than one X chromosome (i.e., 45,X/46,XX; 45,X/47,XXX)—based on the findings of a study that included more than 500 girls with TS. Even when spontaneous puberty occurs, it may not progress normally, and menarche may be delayed or does not happen. Even if menarche occurs, oligomenorrhea and anovulation are common, so that less than 5% of 20-year-old TS women end up having normal menstrual cycles.
Spontaneous pregnancy may occur in 2% to 3% of women with TS. It is more common in women with 45,X/46,XX (or 45,X/47,XXX) cell line mosaicism. However, spontaneous pregnancies occurring in 45,X women, without evidence of mosaicism, are well-documented. In the initial literature on spontaneous pregnancy in TS individuals, increased fetal mortality or malformations were reported. However, this has not been observed in more recent, population-based studies —even for women with X monosomy. If the case where a TS mother has a sex chromosome structural abnormality, such abnormal chromosome may be passed on to her offspring. The risk of maternal complications with spontaneous or assisted pregnancies is quite high for women with TS.
Gonadoblastoma
A gonadoblastoma is a rare benign gonadal tumor, and is almost always found in a (dysgenetic) gonad with Y chromosome material. This tumor has a distinctive histological appearance, which distinguishes it from other gonadal neoplasms. The gonadoblastoma is composed of large germ cells surrounded by sex cord derivatives that resemble small cells with variable granulosa, lutein, or immature Sertoli-like morphology. Because it strongly resembles the histology of a developing gonad, it was assigned the name “gonadoblastoma.” Although the tumor is considered a benign growth, it has both the potential for steroid production (can produce both testosterone and estrogen), as well as for malignant transformation (into a dysgerminoma). Gonadoblastomas are found in approximately 40% of females with 46,XY mixed gonadal dysgenesis. The frequency among girls with TS and Y chromosomal material is between 10% and 30%, but is likely closer to the lower end of this range. These data are based on histologic examination of ovaries that were “prophylactically” removed from girls who carried Y chromosome material. Analyses of Danish health registry data did not find increased morbidity or mortality related to any type of ovarian tumor among women with TS. A large British registry study ascertained gonadoblastoma diagnoses in 8% of women with Y chromosome in their peripheral blood karyotype.
Prophylactic gonadectomy has been standard practice since the time that an excess of gonadal tumors was first observed in girls and women who had both Y chromosome material and intraabdominal gonads. Such a surgical approach was taken because these gonads tended to be nonfunctional, and caused an increased risk for tumor formation. However, spontaneous puberty and even pregnancy may occur in individuals with TS and Y chromosome material. Portnoi et al. reported on an 8-year-old TS girl who was found to have a translocation of Y chromosome material onto the X chromosome (see Fig. 17.2 ). The patient did not undergo gonadectomy, developed spontaneous puberty, and eventually had a well-supervised pregnancy with good outcome. It should also be noted that our clinical experience in TS associated with gonadoblastoma and/or dysgerminoma is based on patients whose karyotype included visible Y chromosome material; in some cases, there even was clinical evidence of virilization. Because of this, the recommendation for ovariectomy was initially only applicable to individuals with visible Y chromosome material or virilization. With the introduction of newer molecular technologies, several reports indicate detection of Y chromosome sequences in 45,X girls without evidence of Y chromosome material on their karyotype. In some of these patients, gonadoblastoma has been detected after gonadectomy. It has been suggested by some that all patients with 45,X monosomy undergo molecular screening for cryptic Y chromosome material. Because it has been shown that PCR amplification may yield false positive results, the risk of overtreatment is a real concern. In addition, immature ovaries may contain nests of germinal cells that are benign but resemble a gonadoblastoma.
There is currently a paucity of information on the use of imaging or serum markers for surveillance of potential gonadal tumors in TS. In any case where Y chromosomes/sequences are detected and gonadoblastoma is suspected, there needs to be education and counseling of patients or family concerning gender identity, sexual functioning, and reproductive consequences relevant to the decision for gonadectomy. Preservation of follicles or oocytes may be an option for some patients undergoing gonadectomy. Further discussion of gonadoblastoma issues is found under the Patients with Y Chromosome section presented later in the chapter.
Cardiovascular System Anomalies
Congenital Cardiovascular Malformations in Turner Syndrome
Congenital cardiovascular malformations (CVMs) are the most serious, life-threatening consequences of X chromosome monosomy. Coarctation of the aorta is one of the first CVMs documented in patients with TS. The frequency and spectrum of TS-specific CVMs, based on data combining chromosomal genotyping and imaging, is summarized in Table 17.2 . Approximately 75% of spontaneously aborted TS fetuses and 30% of living patients have a clinically apparent CVM. Obstructive lesions of the left ventricular outflow tract (LVOT) predominate, ranging in severity from nonstenotic bicuspid aortic valve (BAV) to aortic stenosis, coarctation of the aorta, ascending aortic aneurysm, and mitral valve anomalies (see Table 17.2 ). The most severe form of left-sided CVM, with poor prognosis, is hypoplastic left heart syndrome. The association of TS and left-sided CVM is characteristic among malformation syndromes.
| Mode | BAV | COA | ASD/VSD | PAPVC | ARSC | ETA | Any | |
|---|---|---|---|---|---|---|---|---|
| Gotzsche | Echo | 14% | 10% | 0 | 1% | NR | NR | 26% |
| Mazzanti | Echo | 12% | 7% | 1% | 5% | NR | NR | 23% |
| Volkl | Echo | 18% | 18% | 8% | 5% | NR | NR | 30% |
| NIH | MRA/Echo | 30% | 12% | 1% | 13% | 13% | 49% | 50% |
| Kim | MRA | 39% | 16% | NR | 16% | 6% | 31% | NR |
There exists a significant association between neck webbing and aortic coarctation in girls with TS. This is supported by: (1) observations from aborted fetuses, and (2) additional studies reporting a much higher prevalence of aortic coarctation and BAV in girls with neck webbing. The underlying pathophysiologic mechanism proposes that lymphectasia causes a mechanical defect: engorged fetal lymphatic structures compress the developing ascending aorta, which affects intracardiac blood flow, and ultimately leads to a spectrum LVOT anomalies. Another potential explanation is derived from observations in 45,X fetuses with severe aortic arch defects and aortic valve abnormality. It invokes generalized hypoplasia of the fourth branchial arch tissues associated with defective migration of neural crest cells. LVOT anomaly defects also occur in TS patients without neck webbing, and neck webbing is observed in TS girls with normal cardiovascular structure and function. Taken together, both explanations may play a role here, and haploinsufficiency for certain genes on the X chromosome could lead to both severe fetal lymphedema and aortic heart developmental defects, independent of each other. How manifest the CVM ultimately becomes may be further influenced by genetic variations on the autosomes with/without additional environmental effects.
Nonsyndromic LVOT defects (e.g., aortic coarctation and BAV), in the general population, are not associated with lymphedema or neck webbing. They are also more commonly observed in males than in females by a ratio close to 3:1. This CVM phenotype is observed in individuals with TS who present with either a deletion of just the short arm of the X or Y chromosome. This indicates that loci important for LVOT development are likely present on both Xp and Yp. The putative candidate gene for LVOT development would then have to escape X inactivation in females and be expressed from both the X and Y chromosomes in males. The Yp allele may be more prone to disruption in males, as a consequence of inherent Y chromosome instability. Localization of an LVOT-related gene on the PAR1 of the X chromosome may explain the greater prevalence of LVOT anomalies in males, because the meiotic recombination rate for PAR1 is sevenfold greater in males than females, increasing the overall risk for Yp gene disruption.
Disease of the Aorta
Complications of CVMs are the leading cause of morbidity, as well as premature and increased mortality in TS. The major cardiovascular complications include aortic valve disease and aortic dilation, dissection, or even rupture. Approximately 30% of girls with TS are born with a congenitally abnormal aortic valve. Because careful appropriate screening for the presence of, for example a BAV, is sometimes lacking, congenitally abnormal valves are often not detected until the third decade of life or later. Carolyn Bondy’s group studied 74 patients with BAV, at a median age of 30 years (range 7–67 years). Of these women, 55% had normal valve function, 30% had mild aortic regurgitation, and 15% had moderate to severe aortic regurgitation. In this BAV cohort, aortic stenosis was present in only 2/74 patients. The prevalence of BAV was equal in pediatric and adult subgroups, but adults were more likely to have functionally abnormal valves. A large National Institutes of Health screening study using advanced imaging technology (magnetic resonance angiography or cardiac MRA) confirmed a BAV prevalence of 34% of girls with TS. In a more recent study using similar imaging techniques, Kim et al. report a prevalence of BAV as high as 39%. It is imperative to screen all girls and women with TS for aortic valve pathology, as the aortic valve may deteriorate over time, leading to heart failure. In addition, aortic valve abnormalities are linked to further aorta pathology, with increased risk for the development of aortic dilation, dissection, and rupture.
In individuals who have an abnormal aortic valve, this is often associated with relative dilation of the ascending part of the aorta, using age and body surface area nomograms. The magnitude of ascending aortic dilation is greatest in patients whose valve is also functionally abnormal, although individuals with normal valve function may also develop dilation. In about 10% of patients who have a normal tricuspid aortic valve (and normal blood pressure), mild-moderate ascending aortic dilation can also be found. There is an increasing body of evidence for the existence of a generalized vasculopathy in individuals with TS. Some of the observations supportive of this include an enlarged diameter of the carotid and brachial arteries, as well as aneurysms of other arteries. The term aortopathy has come into wider use and describes the presence of aortic disease of diverse etiologies, including Marfan syndrome and vascular disorders not associated with valvular disease. Aortopathy exists in addition to aortic disease associated with BAV.
From the analysis of epidemiologic data obtained from the Danish registry we can safely state that the risk for serious aortic complications is 100-fold or greater increased in girls and women with TS, compared with the general female population. For example, the median age of aortic dissection was 35 years (in the general female population, this is an event usually observed in the seventh or eighth decade of life), with the few pediatric cases clearly associated with a history of more severe aortic valve disease or coarctation. Aortic valve abnormalities and aortic coarctation, with or without surgical repair, are important predisposing factors for aortic dissection. Hypertension is another risk factor for aortic disease, in the general population, as well as in TS individuals. The presence of aortic dilation is a strong risk factor for aortic dissection in TS, although dissection can occur in women with TS who do not harbor any of the known aforementioned risk factors. Nevertheless, cardiology follow-up in these patients should always include direct and reliable measurements of ascending aortic diameters at the sinuses of Valsalva, sinotubular junction, and ascending aorta for all patients, together with body surface area and age. Spontaneous and assisted pregnancies are associated with a further increased risk for life-threatening aortic complications in TS, and the French College of Obstetrics and Gynecology, as well as the American Society of Reproductive Medicine, have issued stringent guidelines on preconception screening and maternal care aimed at reducing these risks. In the past, too often TS women went through a pregnancy without such detailed evaluations.
Other Cardiovascular Health Issues
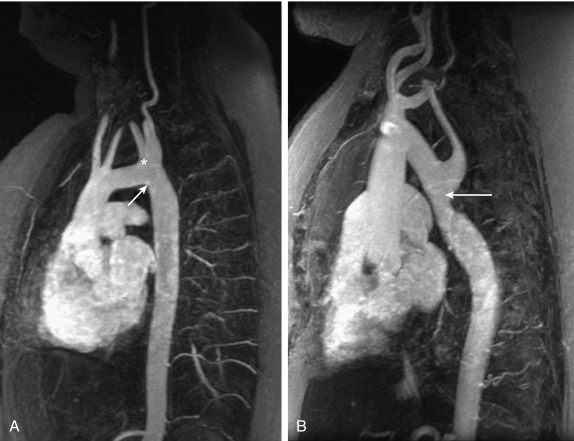
Aortic valve disease and risk for aortic complications are major concerns in TS, but other CVMs are also observed with a higher frequency. Partial anomalous pulmonary venous connection (PAPVC), or return (PAPVR), is increased in TS: it has a reported prevalence between 1% and 16%. The higher prevalence is found after using contrast cardiovascular MRA for screening purposes, and is likely a reflection of the true increased prevalence, because echocardiography is a suboptimal technology to study the anatomy of the pulmonary veins. This diagnosis should especially be pursued in girls with exercise intolerance, unexplained pulmonary issues (reactive airway disease), or evidence of pulmonary hypertension or right heart hypertrophy. The significance of incidentally discovered PAPVC in the screening of asymptomatic patients is unclear. Only 9% to 14% of PAPVC patients reported from two case series had a clinically significant left-to-right shunt necessitating surgical intervention. Aberrant right subclavian artery is another relatively common anomaly (5%–10%; Fig. 17.15 ) that may compress the esophagus, leading to dysphagia and chest pain. One important clinical note relates to the origin of an aberrant right subclavian artery from the descending aorta: this could mask the presence of aortic coarctation if the right upper extremity alone is chosen to screen for upper versus lower extremity blood pressure difference. Atrial or ventricular septal defects and mitral valve disease are more common than in the general female population, but less frequent than aortic defects and PAPVC (see Table 17.2 ).
Minor electrocardiographic findings, including right axis deviation, T wave abnormalities, bundle branch block, QTc prolongation, and increased heart rate variability are more common among girls (30%) and adults (> 50%) with TS, compared with age-matched female controls. Most of these findings are of uncertain significance. Specifically for QTc prolongation, there does not appear to be a worsening of this finding with increasing age. It is recommended to obtain a screening electrocardiogram (ECG) on all patients with TS, and recommend those with a prolonged age-specific QTc to avoid medications associated with QTc prolongation. An ECG should also be done after initiation of QTc prolonging drugs, if these cannot be avoided. However, unlike in familial long QT syndrome, there is no evidence that prolonged QTc in TS is associated with sudden cardiac death. In case right axis deviation is detected on ECG, PAPVC, or PAPVR needs to be considered. Impaired autonomic responses are suggested by resting tachycardia, reduced heart rate variability, and the absence of the expected nocturnal dip in blood pressure. Systemic hypertension is more common in girls and women with TS, compared with age-matched female controls: at least 25% of patients. The higher prevalence of hypertension appears independent of renovascular or aortic disease —although secondary hypertension may occur if vascular pathology or renovascular disease coexists (rare). The hypertension appears not adversely impacted by the usual recommended estrogen treatment regimens. In the majority of patients, the cause of the hypertension is unknown, and it probably has a multifactorial etiology. Sympathetic hyperactivity, increased activity of the renin-angiotensin-aldosterone system, and estrogen deficiency may all be contributory. Further studies are needed to determine ideal systemic blood pressures for girls and women with TS given their smaller stature and increased risk for aortic and also atherosclerotic disease. Moreover, further investigation is required to identify beneficial pharmacotherapy for patients with hypertension or BAV and aortic dilation. We currently define hypertension in TS using the guidelines for the general population and use corrected norms for age, sex, and height when evaluating children.
Cardiovascular Screening in Turner Syndrome
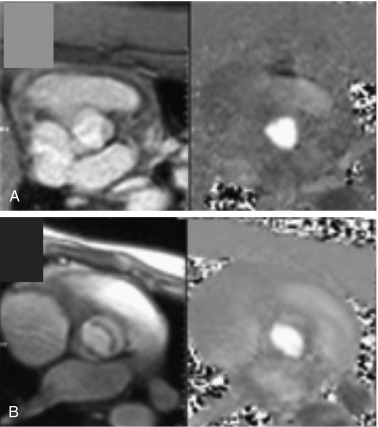
When a girl with TS is diagnosed with a CVM, either in infancy or in childhood, it is recommended she is followed by the pediatric cardiology team, ideally at a specialized tertiary care center, and preferably by a cardiologist with special expertise in following congenital heart disease. Most girls diagnosed with TS later in childhood or adolescence do not present with clinically obvious cardiovascular disease. Close cardiovascular examination and consultation with pediatric cardiology are required at the time of diagnosis however, with particular attention to blood pressure measurements in all extremities. The mode of imaging and the frequency of follow-up are determined by the clinical situation in each case. Transthoracic echocardiography has been the standard approach to screen for congenital CVM for decades, but this modality has important limitations related to operator dependency and limited acoustic windows, especially in the context of abnormal chest anatomy and obesity, which are common physical features seen in TS. Cardiovascular magnetic resonance (CMR) imaging provides excellent visualization of cardiovascular anatomy and function, with dimensions of ascending, transverse, and descending aorta, in any desired imaging plane (see Fig. 17.15 and Fig. 17.16 ). Importantly, CMR may detect significant cardiovascular anomalies, including chronic aortic dissection, aortic coarctation and dilation, aortic valve abnormality, and PAPVC (PAPVR) not always so easily demonstrated by routine echocardiography.
Asymptomatic BAV or aortic dilation and some cases of coarctation pose risks for aortic complications, but may escape detection with standard evaluation and echocardiography. Current recommendations include the more advanced imaging (i.e., CMR) when possible, without excessive risks of sedation or radiation exposure, usually by age 12 years. The detection of asymptomatic BAV and aortic dilation is crucial to assess the individual’s risk for aortic complications, as well as the requirement for ongoing cardiac monitoring during adulthood. In addition, since 2017 new guidelines are available that help educate the young patient and her family about the risks of high-impact or high-resistance exercise and the knowledge of coexisting CVMs will enhance counseling in this area.
Ongoing Cardiac Care
Pediatric TS patients with known congenital CVMs require continued care by the pediatric cardiology team and should be transferred to adult congenital heart disease specialists when appropriate for age. Patients who have had aortic coarctation repair in childhood need ongoing surveillance for detection of hypertension, development of aortic valve disease, as well as imaging of the aorta to monitor for restenosis, dilatation, or true aneurysm formation. Patients with BAV or aortic dilation need regular and continued monitoring for hypertension, valve function, and potential progression of aortic dilation. The optimal frequency of monitoring is not exactly known because it is uncertain if there is continuous progressive dilation of the aorta, as seen in Marfan syndrome, or whether changes occur by leaps rather than by gradual transition. The latter process could be influenced, for example, by a stressor, such as a motor vehicle accident, participation in high-resistance exercise, or an associated pregnancy. Available evidence describes a small annual rate of dilation near the detection limit of current imaging methods. Further studies are required to identify parameters that may be more useful in predicting imminent aortic decompensation, such as aortic wall thickness, compliance, or circulating markers (e.g., natriuretic peptide). The aforementioned 2017 guidelines provide a cardiac imaging and cardiology evaluation follow-up protocol, which was arrived at by reviewing the current available literature, combined by consensus agreement from experts in the field of TS cardiac disease.
Beta-blockers and angiotensin receptor blockade have shown some efficacy in preventing aortic dilation in Marfan syndrome, but neither form of treatment has been carefully investigated in affected TS patients. Children at increased risk for aortic dissection because of BAV and aortic dilation, with or without hypertension, and their families should be counseled about possible presenting symptoms (e.g., chest or back pain). Wearing a medical identification bracelet indicating the presence of aortic disease could be of benefit in case the family is not able to advocate for their child in distress. Delayed or missed diagnosis is the most important cause of fatality in acute aortic dissection, a major concern if the emergency department staff fails to consider dissection in an atypical patient presenting with clinical features consistent with acute aortic dissection. The most common presenting symptom should be pain, often of abrupt onset. Typically, the pain is increasing in intensity and may be described as sharp. The pain is typically felt in the anterior chest but may be experienced in the back in case of descending aorta dissection.
For the patient with no identified cardiovascular defects after adequate cardiovascular evaluation and imaging, routine pediatric care with continued monitoring of blood pressure is advised. It seems also prudent to reevaluate aortic dimensions at 5- to 10-year intervals (see later). Heart-healthy moderate aerobic exercise is emphasized and should be encouraged. As noted before, eligibility for competitive sports for all with TS should be determined by a cardiologist, after a comprehensive evaluation that includes magnetic resonance imaging (i.e., CMR) of the aorta.
Risk for Premature Atherosclerotic Disease
Epidemiologic analysis of registry data from Denmark described a 3-fold increased risk of coronary and cerebrovascular disease among women with TS. It was not determined if the coronary disease and stroke were secondary to atherosclerosis or to underlying congenital CVMs. Adults with nonsyndromic aortic coarctation have an increased risk of hypertension, coronary disease, and stroke—despite apparently successful coarctation repair. Total cholesterol concentrations are often elevated in untreated girls with TS, compared with age-matched females, with improvements in the metabolic profile with GH treatment. Atherogenic risk factors, including abdominal adiposity and lipid profile, are higher in women monosomic for the maternal X chromosome versus those with retained paternal X chromosome, but they are not usually increased into the atherogenic range. Recommendations for preventing atherosclerosis apply equally to girls with TS and the general pediatric population and include promoting a healthy lifestyle, monitoring blood pressure, and preventing obesity. Although many pediatricians initiate lipid screening around 10 years of age, with repeat assessment in the transition years (between ages 17 and 21 years), the recent guidelines recommend a lipid profile to be performed in individuals who have at least one risk factor for cardiovascular disease and starting at 18 years.
Renal Anomalies
Some 20% to 30% of girls with TS will have a renal abnormality. Duplication of the pelvocaliceal collecting system, a horseshoe kidney, renal ectopia, and unilateral renal agenesis are the more commonly occurring anomalies. The etiology of the renal anomalies is unknown. There is no apparent correlation between the renal anomalies and other features of TS, such as neck webbing or congenital CVMs. However, a horseshoe kidney is thought to occur more commonly in patients with a 45,X karyotype. Most TS children with congenital renal anomalies have normal renal function, but some are at increased risks for urinary tract infections. Pyelonephritis can occur if there is evidence of obstructive collecting system lesions, and this may require surgical repair. It is recommended that all patients undergo ultrasound imaging of the renal system at diagnosis. Additional tests (e.g., voiding cystourethrogram or renal scintigraphy) may be required in patients with (recurrent) urinary tract infections. Renal function should be tested on a regular basis for those individuals with a horseshoe or single kidney. Additional studies are necessary to determine if renal anomalies diagnosed in TS patients are associated with gradual worsening of renal function or the development of proteinuria and hypertension during later life.
Otologic Disorders
Recurrent middle ear infections are one of the most common medical problems in young girls with TS. Anderson et al. first described the presence of middle ear pathology in girls with TS: recurrent otitis media, eardrum perforations frequently requiring surgical intervention, and significant hearing loss occurring in up to one-quarter of affected patients. Many other reports further address the conductive hearing loss that occurs secondary to refractory otitis media in TS, and having a karyotype with loss of the short arm of the X chromosome (Xp) seems to predispose to this. Many girls will require repeated tympanostomy tube placement. Despite this, some patients may develop complications, including the development of cholesteatoma. The frequent otitis media may be the consequence of abnormalities in the shape and growth of the cranial base, leading to an abnormal relationship of the middle ear to the Eustachian tube. Together with anatomic abnormalities of the palate, a predisposition to fluid collection and secondary infection may be predisposing factors explaining the common occurrence of this problem, rather than a form of immunologic dysfunction, as initially entertained.
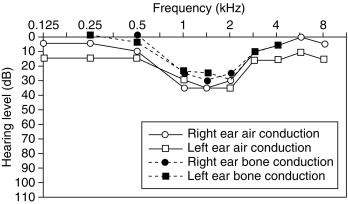
Although conductive hearing loss is most common and severe in children and is correlated with middle ear pathology, sensorineural hearing loss is more common in adults with TS. This sensorineural hearing loss is usually bilateral and characterized by a dip in the mid-frequency range (between 500 and 2000 Hz) during audiology testing ( Fig. 17.17 ). Sensorineural hearing loss seems progressive and is likely not a pure congenital abnormality. By middle age, more than 90% of women with TS have a hearing loss of more than 20 dB, and more than 25% will eventually require hearing aids. Significant conductive hearing loss in adults with TS may coexist, either as a consequence of scarring of the tympanic membrane, as a result of the frequent bouts of otitis during childhood, or it may indicate ongoing middle ear pathology. The aggressive treatment of ear, nose, and throat problems in childhood to avoid injury to the inner ear may reduce the risk of long-term hearing loss.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


