A third interpretation of the role of haploinsufficiency in tumorigenesis is based on the exquisite sensitivity of cells to even small changes in the levels of some cancer-associated proteins. Thus, a 50% functional reduction in a TSG product may be sufficient to endow a cell with a relative advantage for proliferation. Experimental evidence for this dosage-sensitivity effect has been shown for several TSGs, including TP53, BRCA1, BRCA2, and PTEN. For example, a subset of tumors arising in p53 +/− mice, or in patients with Li-Fraumeni syndrome, retain the wild-type allele, suggesting that haploinsufficiency of TP53 may be sufficient for tumor initiation. 28–30 Finally, the pro-tumorigenic effect imparted by haploinsufficiency might also be dependent on the loss or gain of function of other alleles. An example of this interaction is illustrated in mouse models of Pten and Tp53 deficiency. Haploinsufficiency of Pten in the context of wild-type p53 enhances proliferation and subsequent transformation of prostate epithelial cells. In contrast, complete loss of Pten in this tissue triggers a p53-dependent senescence program, with tumors arising only after Tp53 inactivation. 8
Interconnecting the pRB, p53, and mTORC1 Pathways
Similar to an evolutionary process driven by natural selection, the acquisition of the malignant phenotype can be best described as an iterative process of somatic mutation followed by clonal expansion. The current consensus is that normal cells must acquire at least four distinct mutational or epigenetic events (including gain- and loss-of-function alterations) in order to bypass proliferative control. These events alter critical signaling networks that ultimately control the decision of a cell to proliferate, senesce, or die. The discovery of these pathways represented a major success in the history of cancer biology, for it made possible the assignment of a growing number of cancer-associated gene products to a much more limited number of interacting networks. 2 Because of their high association with human malignancies, a more detailed description of pathways centered on pRB, p53, and mTORC1 is used in the next sections to illustrate the emergent complexities of tumor suppression. Whenever necessary, the relevant nodes interconnecting these pathways are stressed.
The pRB Tumor Suppressor Pathway
Despite the low incidence of RB-1 mutations, it has become clear that deregulation of the pRB pathway is present in most (if not all) human cancers. Deregulation can occur through gain or loss of function of various components of the pathway. In most cases, these alterations ultimately result in pRB phosphorylation, a modification necessary for G1-S cell cycle transition. 31,32 In G1, hypophosphorylated (active) pRB forms repressive complexes with E2F transcription factors at gene promoters of S-phase genes, a function in part mediated by the recruitment of histone-modifying complexes to these sites. These modifications result in a silent chromatin configuration that effectively turns off S-phase genes. Signals that promote proliferation must reverse this inhibition, and they do so through pRB phosphorylation. 19 In mammalian cells, pRB is phosphorylated by cyclin-dependent kinases (CDKs), a group of enzymes that require the binding of short-lived proteins called cyclins in order to become active. As shown in Figure 3-1 , CDK4 and CDK6 form active complexes with D-type cyclins in early G1, whereas CDK2 is activated by E-type cyclins in late G1 and S phase. Following phosphorylation, hyperphosphorylated (inactive) pRB releases E2F transcription factors, which leads to derepression of S-phase genes (see Figure 3-1). Predictably, cyclins and CDKs act as oncoproteins in human cancers. For example, CCD1 (encoding cyclin D1) is amplified or overexpressed in more than 50% of human breast cancers. 31,33
CDKs are also subject to negative regulation by two families of CDK inhibitors. Members of the INK4 family, such as p16INK4a, bind to CDK4 or CDK6 and prevent their association with D-type cyclins. On the other hand, members of the CIP1/KIP1 family, which include p21CIP1 and p27KIP1, form inhibitory complexes with CDK2 and cyclin E. 31 In both cases, CDK inhibition results in pRB activation and cell cycle arrest (see Figure 3-1). As expected, many CDK inhibitors behave as tumor suppressors in human malignancies. For example, CDKN2A, the gene encoding p16INK4a, is frequently deleted or epigenetically silenced in cases of familial melanoma and in several sporadic tumors. 34
The CDKN2A-containing locus is also remarkable in that it encodes a second, structurally and functionally unrelated protein from an alternative reading frame and a different gene promoter. 35 This alternative reading frame protein (ARF, also called p14ARF in humans and p19Arf in mice) stabilizes p53 by directly inactivating MDM2, the E3-ubiquitin ligase that targets p53 for degradation. Therefore, loss-of-function mutations affecting ARF, as well as gain-of-function mutations in MDM2, can both have a similar destabilizing effect on p53. ARF-specific inactivating mutations have been described in a subset of human melanomas; MDM2 amplifications, on the other hand, are common in human sarcomas. 2 Notice that the unique genomic arrangement of the CDKN2A locus, encoding both p16INK4a and p14ARF, means that deletions of this locus would simultaneously compromise the functions of the pRB and p53 pathways. Hence, in a manner reminiscent of the inactivation of pRB and p53 by the SV40 large T antigen, cells harboring certain CDKN2A mutations display an enhanced proliferative capacity in vitro, which in many cases is sufficient for the establishment of continuously proliferating (immortal) cell lines. This immortalization step, in turn, sensitizes cells to oncogene-mediated transformation. 36
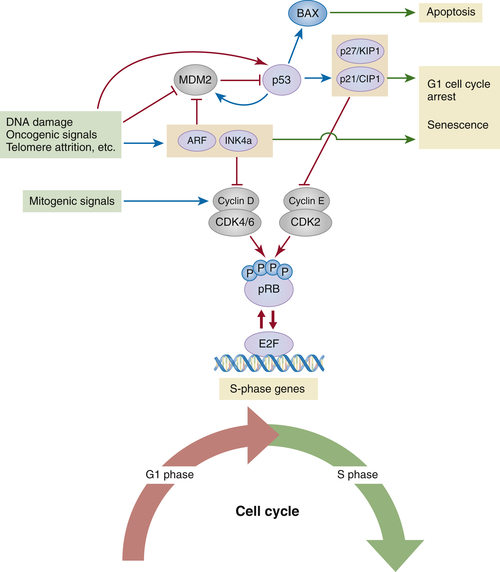
Figure 3-1 The pRB and p53 tumor suppressor pathways Under physiological conditions, signals that promote proliferation (i.e., growth factors) induce the expression of D-type cyclins, short-lived proteins required for the activation of CDK4 and CDK6 (CDK4/6) in early G1. In cooperation with cyclin E–CDK2 complexes, cyclin D1–CDK4/6 complexes contribute to the phosphorylation-mediated inactivation of pRB and the derepression of E2F-responsive (S-phase) genes. A variety of stresses, including DNA damage, telomere erosion, and oncogenic stress, turn on signaling cascades that activate the CDKN2A locus. ARF, one of the tumor suppressors produced from this locus, inhibits MDM2 (the E3-ubiquitin ligase that targets p53 for degradation), resulting in the stabilization of p53 and the induction of p53-dependent transcriptional programs (the transcriptional activity of p53 can also be affected by numerous posttranslational modifications that, for simplicity, are not depicted here). Depending on the cell type and/or the nature and magnitude of the stress, p53 can induce the expression of genes that promote apoptosis or, alternatively, genes involved in cell cycle arrest, such as the gene encoding the CDK inhibitor p21CIP1. p21CIP1, in turn, inhibits cyclin E–CDK2 complexes and leads to pRB-dependent cell cycle arrest. The second tumor suppressor encoded by the CDKN2A locus, p16INK4a (INK4a), is also induced in response to stress (with or without concomitant induction of ARF), most typically oncogenic-dependent stress. By antagonizing CDK4/6, p16INK4a also activates pRB and prevents entry into S phase. Red arrows and red T-shaped connectors indicate activating and inhibitory processes, respectively; blue arrows indicate transcriptional induction; green arrows point to final cellular outcomes.
The p53 Tumor Suppressor Pathway
As already mentioned, p53 is a multifunctional transcription factor that is activated in response to a variety of stress conditions, including DNA damage, activated oncogenes, telomere shortening, spindle damage, hypoxia, and metabolic stress. 7 Depending on the cell type and the magnitude or nature of the stress, the transcriptional program activated by p53 can lead to one of three outcomes: cellular senescence, G1/G2 arrest, or apoptosis (see Figure 3-1). Mirroring the diversity of stress signals is the equally diverse number of intracellular networks involved in the transmission of these signals to p53. What all these networks have in common, however, is the ability to activate p53 via posttranslational modifications (including phosphorylation, acetylation, methylation, and sumoylation) that enhance p53’s capacity to transcribe target genes and also increase the half-life of the protein. We have seen already that one way to stabilize p53 involves the ARF-dependent inhibition of MDM2.
Among numerous p53 targets, the gene encoding p21CIP1 (CDKN1A) is probably the best-known example that connects p53 to the pRB pathway (p53-mediated upregulation of p21CIP1 leads to CDK inhibition and pRB-mediated cell cycle arrest). In addition to targets involved in cell cycle arrest, p53 also induces genes that control or promote apoptosis (such as BAX, PUMA, and PIG3), a cellular outcome favored by some cell types undergoing extensive DNA damage. 37
Given the pleiotropic roles of p53 in tumor suppression, it is not surprising that mutations in TP53 are almost a universal feature of human cancers. Most of these mutations (∼74%) are missense mutations that fall within the DNA-binding domain and therefore disrupt p53’s ability to bind cognate promoter sequences on target genes. 22
pRB and p53 Pathways in Action: Cellular Senescence
The connection between the two major tumor suppressors, pRB and p53, and cellular senescence is a recurrent theme throughout this chapter. Because of the growing awareness that cellular senescence constitutes a physiological barrier against tumor initiation and progression, a brief description of two related variants of senescence, and their connection with the pRB and p53 pathways, is necessary (see Figure 3-1).
Cellular senescence is a form of irreversible cell cycle arrest associated with a unique gene expression profile and distinctive cell morphology. 38 The first description of this phenomenon can be traced back to efforts to propagate human cells in vitro. Explanted human cells typically proliferate for a variable period of time but eventually undergo “replicative” senescence in response to the attrition of telomeres (the protective chromosomal termini) that accompanies each cell division. Telomere disruption triggers a stress response that in many respects is identical to a p53-dependent DNA damage response (DDR). 39 Before the signs of DDR are evident, however, an early induction of p16INK4a also contributes to limit the proliferative capacity of human cells, which is illustrated by the significant delay in the onset of replicative senescence observed in p16INK4a-deficient cells. Nonetheless, in order to completely bypass senescence, human cells must also overcome the p53 pathway and reactivate telomerase (the enzyme responsible for telomere maintenance). 38,40
A form of replicative senescence associated with the induction of p16INK4a and p53 is also observed in primary mouse cells, but, in contrast to human cells, the inactivation of just one of these tumor suppressors suffices to bypass senescence. Because murine cells constitutively express telomerase, senescence in this case might be a response to nonphysiological culture conditions (i.e., high oxygen levels). 38
In addition to replicative senescence, primary cells also undergo “premature” senescence in response to oncogenic stress (i.e., overexpression of activated HRAS), a phenomenon known as “oncogene-induced senescence” (OIS). Similar to replicative senescence, OIS is also dependent on the induction of p16INK4a and p53, although the relative contribution of these tumor suppressors varies between species and cell types. 38,41 Recently, OIS has been confirmed as a barrier to tumorigenesis in vivo. For example, benign melanocytic tumors called “nevi” are associated with activating mutations in the oncoprotein BRAF. These lesions are typically positive for markers of senescence, including elevated levels of p16INK4a. As expected, loss of p16INK4a in these lesions accelerates the formation of malignant melanomas. 42,43
The mTORC1-Dependent Pathways
Alterations in a cell’s ability to respond to metabolic and growth-promoting signals constitute common features of cancers. At the center of these regulatory networks is mTORC1 (mammalian target of rapamycin complex 1), a kinase complex that integrates nutrient and growth factor availability with downstream effectors involved in cell growth and proliferation. mTORC1’s main function is to promote biosynthetic processes (i.e., protein synthesis and ribosome biogenesis) that increase cell mass. Two pathways that converge on mTORC1 (the PI3K- and AMPK-dependent pathways) are particularly relevant to cancer 44,45 (Figure 3-2 ).
Activation of tyrosine kinase receptors, most classically insulin-like growth factor receptor (IGFR), activates the lipid kinase PI3K (class I phosphatidylinositol-3-kinase), an enzymatic complex that catalyzes the production of phosphatidylinositol-3-phosphate (PIP3). The local increase in PIP3 is in turn required for the activation of AKT kinases. AKTs inhibit the GTPase activating complex TSC (tuberous sclerosis complex, composed of TSC1 and TSC2 subunits), a process required to keep Rheb (a small GTPase) in its GTP-bound (active) conformation. As a result, AKT-mediated inactivation of TSC increases the pool of active Rheb, which in turn enables mTORC1 activation (see Figure 3-2). AKT-mediated phosphorylation of substrates other than TSC can also affect proliferation by controlling pRB activity. These substrates include the FoxO (Forkhead box O) family of transcription factors and GSK3-β (glycogen synthase kinase 3-beta). Under basal conditions, FoxOs induce the expression of the CDK inhibitors p21CIP1 and p27KIP1, whereas GSK3-β participates in the degradation of cyclin D1 (see Figure 3-2). Therefore, AKT-mediated inactivation of FoxO and GSK3-β reduces the levels of CDK inhibitors and increases the levels of cyclin D1, outcomes that cooperate to promote cell cycle progression (see Figure 3-2). 46
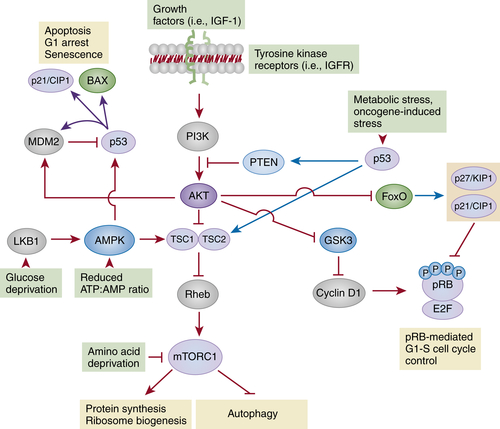
Figure 3-2 The PI3K-AKT-mTORC1 and LKB1-AMPK-mTORC1 pathways The main function of mTORC1 is to regulate biosynthetic pathways involved in cell growth, cell survival and proliferation. The proper function of mTORC1 depends on its ability to integrate inputs from two pathways: the PI3K-AKT axis, involved in the transduction of growth-promoting signals and the LKB1-AMPK axis, which monitors the energy status of the cell. Ligand-mediated activation of tyrosine kinase receptors (i.e., insulin-like growth factor receptor, IGFR) activates PI3K, which increases the local concentration of the lipid second messenger phosphatidylinositol-3-phosphate, PIP3. This activity is counteracted by PTEN, a lipid phosphatase that removes phosphate groups from PIP3. PI3K-mediated increase of PIP3 levels activates the AKT family of kinases, which inhibit the GTPase activating complex TSC (TSC1-TSC2). Inhibition of TSC allows Rheb to remain in its active (GTP-bound) conformation, resulting in mTORC1 activation. In addition, AKT-mediated phosphorylation of FoxO transcription factors and GSK3-β can also lead to pRB inactivation and cell cycle entry by reducing the expression of CDK inhibitors (p21CIP1 and p27KIP1) and increasing the stability of cyclin D1. The AMPK complex and its upstream activator LKB1 monitor the intracellular levels of glucose and ATP and modify the functional status of mTORC1 accordingly. In situations of metabolic stress, the resulting increase in the AMP:ATP ratio leads to the activation of LKB1, which phosphorylates and activates AMPK. Active AMPK can then inhibit mTORC1 through TSC-dependent and TSC-independent mechanisms. As shown here, in response to metabolic stress, p53 can directly induce the expression of TSC2 and PTEN, contributing to mTORC1 inhibition. Conversely, AMPK can also phosphorylate and activate p53. Red arrows and T-shaped connectors indicate activating and inhibitory processes, respectively; blue arrows indicate transcriptional induction.
Normal cells must also implement mechanisms to reduce the PI3K-AKT activity in order to adjust the rates of protein synthesis to the available growth factors. One important mechanism of inhibition involves the activation of PTEN (phosphatase and tensin homologue), a lipid phosphatase that catalyzes the dephosphorylation of PIP3 and thus counteracts PI3K activation (see Figure 3-2). 44
In cancer cells, activation of the PI3K-AKT-mTORC1 pathway can occur through several mechanisms. These include aberrant activation of tyrosine kinase receptors (TKRs), activating mutations in PI3KCA (the gene encoding the p110α subunit of PI3K), amplification of AKT1, downregulation of TSC2, or loss-of-function mutations in PTEN. 46 Loss of PTEN in particular constitutes a remarkably frequent alteration in human malignancies. Similar to other tumor suppressors, germline mutations in PTEN are also associated with inherited cancer syndromes (including Cowden disease), and hemiallelic loss of Pten in mice results in tumors arising in multiple epithelial tissues, including the intestine, prostate, and mammary gland. However, unlike other tumor suppressors, biallelic deletion of Pten can activate a p53-dependent senescence program that opposes transformation, suggesting that PTEN is an obligate haploinsufficient tumor suppressor. By comparison, mutations affecting other negative regulators of the PI3K-AKT-mTORC1 pathway are rare events in sporadic tumors, although germline deletions of TSC1 or TSC2 are known to be associated with cancer-prone syndromes. 44
The second signaling pathway that modulates mTORC1 is the AMP-activated protein kinase (AMPK) pathway. During periods of nutrient deprivation, AMPK is activated by at least two mechanisms: (1) increased levels of AMP (due to a drop in ATP production) and (2) phosphorylation by LKB-1, a kinase that is itself activated in response to metabolic stress (i.e., glucose reduction) (see Figure 3-2). Active AMPK phosphorylates TSC2, leading to the activation of the TSC complex, inactivation of Rheb and mTORC1, and the consequent inhibition of protein synthesis and cell growth. 44 An important consequence of mTORC1 inhibition is a shift toward a predominantly catabolic metabolism. This is in part achieved through the induction of autophagy, the process in which organelles and protein complexes are targeted to the lysosome for degradation. 47
It follows from this account that cancer cells must overcome the AMPK-dependent checkpoint in order to sustain proliferation under suboptimal metabolic conditions. Among the mechanisms of AMPK inactivation, loss of function of LKB-1 is probably the best known. Inactivating mutations of the gene encoding LKB1 (STK11) have been identified in patients with Peutz-Jeghers syndrome, a condition that predisposes individuals to the development of several types of cancer. Recently, STK11 was also found mutated in sporadic cases of non–small-cell lung and cervical carcinomas. 44
In recent years, it has become evident that most alterations in cancer-associated genes can result in metabolic changes that involve mTORC1-dependent pathways to variable degrees. In particular, the role of p53 in mediating adaptation to metabolic stress is becoming increasingly evident. Thus, inhibition of the AKT-mTORC1 axis, as well as activation of AMPK, can both lead to the induction of p53. The first mechanism is a consequence of the ability of AKT to activate MDM2. Therefore, a reduction in AKT function (i.e., secondary to PTEN activation) activates p53 by removing the negative regulation imposed by MDM2. On the other hand, in situations of nutrient deprivation, AMPK-mediated phosphorylation of p53 increases its half-life and transcriptional activity. 48
Epigenetic Modifications and Tumor Suppression
Evidence accumulated in the past decade has led to the realization that epigenetic changes affecting oncogenes and TSGs constitute important events contributing to the hallmarks of cancer. The cancer “epigenome” is characterized by global and gene-specific heritable modifications that affect gene expression but do not involve changes in DNA sequence. Three types of cancer-associated epigenetic modifications are currently recognized: DNA methylation, histone modifications, and micro-RNA (miRNA)-mediated gene silencing. 49 Although they are discussed separately in this chapter, we should emphasize that these are functionally interdependent mechanisms. For example, some miRNAs can modify the epigenetic landscape by regulating the expression of proteins involved in histone modifications or DNA methylation. Conversely, miRNA expression itself can be altered by DNA methylation or histone modifications (Figure 3-3 is a summary of the most relevant epigenetic changes observed in cancer cells).
DNA Methylation
DNA methylation is the covalent addition of a methyl group to the cytosine ring of a CpG dinucleotide. Although CpG dinucleotides are widely distributed throughout the genomes of eukaryotic organisms, CpG-rich regions (also called CpG islands) are particularly enriched in the promoter regions of genes. The silencing effect of DNA methylation can result from the direct inhibition of transcription factor binding to promoter regions or from the recruitment of repressive protein complexes to methylated regions. As explained later in this chapter, this latter mechanism often results in a more condensed, transcriptionally silent chromatin configuration. 49
Genome-wide hypomethylation and promoter-specific hypermethylation are common features of cancer. These alterations can be detected in benign lesions and early-stage tumors, suggesting that they may precede classical genetic events. Global loss of DNA methylation at CpG islands was the first epigenetic alteration identified in cancers. In particular, hypomethylation at repetitive sequences in the genome is associated with genomic instability. 50,51 However, the most recognized epigenetic alteration in cancer cells is the promoter hypermethylation affecting TSGs. One of the first examples of this silencing mechanism was discovered in some RB1 alleles associated with retinoblastoma. 50 Since then, hypermethylation as a mechanism of gene inactivation has been demonstrated for other TSGs, including INK4a/p16 (CDKN2A), MLH1, BRCA1, VHL and CDH1 (encoding E-cadherin). In many cases, collaboration between hypermethylation and genetic inactivation can also be documented. For example, promoter hypermethylation contributes to the inactivation of the wild-type allele of CDKN2A in colorectal cancer cells that have already lost one allele through deletion. That hypermethylation is causally involved in the repression of TSGs has been confirmed through the use of demethylating procedures. For example, acute elimination or inhibition of DNMT1 (a DNA methyltransferase) in colon cancer cells is sufficient to reactivate INK4a/p16 and induce cell cycle arrest. 51
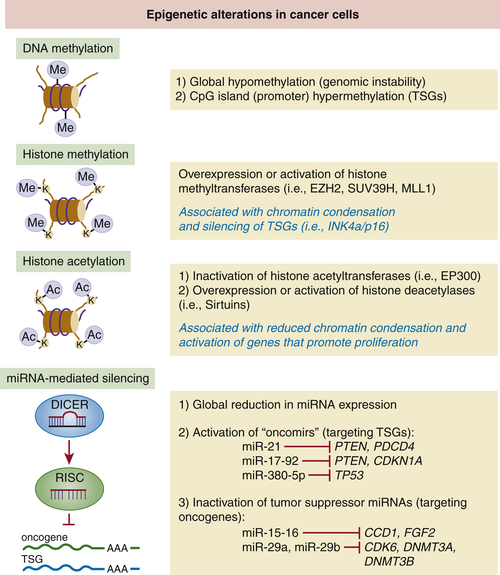
Figure 3-3 Epigenetic mechanisms involved in tumor suppression A brief summary of the main epigenetic changes affecting cancer cells.
Histone Modifications
The second group of cancer-associated epigenetic changes involves the covalent modification of histones, the proteins that form the core of nucleosomes. Among numerous modifications, acetylation and methylation of lysine (K) residues at the N termini of histones H3 and H4 are probably the best known. It has been proposed that the combinatorial addition or removal of acetyl and methyl groups to several K residues of H3 and H4 may serve as a “histone code” that dictates the degree of chromatin condensation and, therefore, the extent to which a genomic locus becomes transcriptionally active. For example, trimethylation of lysines 4, 36, or 79 and acetylation of lysines 9 or 14 of histone H3 are both associated with a relaxed chromatin configuration that facilitates transcription. Conversely, di- or trimethylation of lysine 9 or 27 of histone H3 is associated with a more condensed, transcriptionally silent chromatin configuration. It is important to keep in mind that the enzymatic complexes involved in these modifications (see later discussion) cooperate extensively with DNA methyltransferases (the enzymes that catalyze DNA methylation) to produce stable chromatin states. 51
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


