Solid Tumor Cytogenetics: Current Perspectives
Department of Pathology and Laboratory Medicine, David Geffen School of Medicine, University of California Los Angeles, 2-226 Rehab Center, 1000 Veteran Avenue, Los Angeles, CA 90024, USA
Keywords
• Cytogenetics • Chromosome • Solid tumor • Fluorescence in Situ Hybridization (FISH) • Prognosis • Diagnosis • Clinical • Review
Following the discovery in 1956 that the human complement comprises 46 chromosomes (23 pairs of autosomes, XY sex chromosomes),1,2 the development of human cytogenetics has been one of continuous discovery with impressive advances in the underlying methodologies. The translocation t(9;22) or the Philadelphia chromosome identified by conventional cytogenetics in chronic myeloid leukemia in 19603 and the ERG-TMPRSS2 fusion transcript identified by high-throughput expression analysis in prostate cancer in 20094 are the paradigms of this remarkable journey and technological advances. The limitation of conventional cytogenetics (requirement of dividing cells and limited resolution) was the driving force to develop alternate molecular methods that enable the analyses of nondividing cells along with offering better genome resolution. Fluorescence in situ hybridization (FISH) was the first such molecular method to be developed,5 followed by several others techniques such as Multicolor FISH (mFISH)/Spectral Karyotyping (SKY), metaphase comparative genomic hybridization (mCGH), Bacterial Artificial Chromosome (BAC), or oligonucleotide/single-nucleotide polymorphism (SNP)-based comparative genomic hybridization (aCGH). All of these molecular methods are based on the principles of in situ hybridization in which single stranded complementary sequences of DNA are hybridized to genomic targets under appropriate conditions to form stable hybrid complexes, which are then visualized usually by a fluorescence detection system.6–8 As expected, with the advent of every new methodology, the role of conventional karyotyping in routine clinical practice is debated and its utility questioned. Despite its recognized limitations, conventional cytogenetics in conjunction with FISH continues to remain an important and integral component in the diagnosis and management of neoplastic conditions. The ability to effectively detect the vast majority of clinically relevant chromosomal aberrations with a rapid to acceptable turnaround time makes chromosome analysis the most cost-effective detection/screening tool currently available in modern pathology.
Over the past 4 decades, the karyotypes of more than 60,000 neoplasms have been published.9 Because of the ease with which chromosome preparations can be obtained, leukemias lead the way, with lymphomas and solid tumors following behind. Conventional chromosomal analysis has been extremely crucial in establishing the principle that cancer is a genetic disease and that tumor development is a multistep process driven by accumulation of microscopic and molecular genomic changes. The recognition of recurrent chromosomal changes in specific disease entities has been the basis for the reclassification of many hematopoietic and solid tumors by the World Health Organization (WHO). Over the past decade, an increasing number of solid tumors have been rigorously analyzed by molecular methods, and these studies have revealed additional novel chromosomal alterations (translocations, deletions, gains, and amplifications). Routine karyotyping of solid tumors is still performed in many laboratories but, for clinical purposes, many of the genomic changes identified by conventional and molecular methods are primarily detected by DNA-based FISH assays. It is more sensitive and specific, has a rapid turnaround time, allows for a direct correlation with morphology/histology, and the analyses can be performed both retrospectively and prospectively on a variety of tissue types. An improvement in the quality of FISH probes, microscopy, and standardization of fluorescent signal guidelines has allowed routine FISH testing in formalin-fixed paraffin embedded sections. However, the protocols must be validated in individual laboratories, and parameters such as fixative times, section thickness, quality of the tissue preservation, probe hybridization conditions, areas of malignant cells, and number of abnormal cells scored must be appropriately considered prior to reporting, as these factors are known to affect results and, consequently, clinical management. Commercial FISH probes are now available for all the major rearrangements, and amplifications of clinically relevant genomic regions and new probes or multicolor probe panels continue to be developed. Given the usefulness of both the specific and the global chromosomal profile, our laboratory routinely performs conventional chromosomal analysis and/or FISH on sarcomas and renal cell carcinomas. The other solid tumors are primarily assayed by FISH.
Methodological Considerations
Most malignant solid tumors exhibit chromosomal abnormalities. The success of obtaining an abnormal karyotype depends to a large extent on the pre-analytical and culture protocols adapted by the laboratory.10 Factors that contribute to failure or only normal karyotypes (from non-malignant cells) include: (1) microbial contamination—this is very common and generally occurs when specimens are placed on unclean cutting boards when dissecting the tumors, use of nonsterile blades or transport media without antibiotics, or poor handing of the specimens prior to setting up the culture. Tumor specimens from regions that are inherently colonized by bacteria (eg, gastrointestinal tract, lung, and cervix) require special attention; (2) tumor necrosis—many solid tumors are composed of necrotic or fibrotic regions containing very few viable cells or cells useful for short or long-term culture. For cytogenetic analysis, it is crucial to select a maximally viable tumor region(s); (3) overgrowth of neoplastic cells by non-neoplastic or reactive cells—most solid tumor biopsies, depending on the type, stage, and location of the tumor, contain a mixture of neoplastic and non-neoplastic cells (fibroblasts, normal epithelial cells, or endothelial cells). If the culture is not monitored and harvested in a timely manner, the non-neoplastic cells usually overgrow and, sometimes, inhibit—by using up resources and space—the slower growing tumor cells. This is the most likely explanation for a normal-diploid karyotype in solid tumor cytogenetics; and (4) unpredictable growth of neoplastic cells in vitro—cancer cells often do not recapitulate the in vivo growth (doubling time) pattern in vitro. Many solid tumors, particularly those of epithelial origin, are difficult to grow in culture and require optimal culture conditions (seeding density, media, and growth factors), and it is prudent to be aware of the appropriate conditions required by the different tumor types. In general, tumors of soft tissue and bone, renal cell carcinomas, and certain benign tumors harboring recurrent chromosomal alterations are relatively easy to grow; therefore, the cytogenetic data in these tumors predominate in the literature. For certain cancers like ocular melanoma, fine-needle aspirate (FNA) approaches with the cells smeared on slides and their analyses by targeted FISH probes are very useful.11
Chromosomal Changes In Solid Tumors: An Overview
The vast majority of chromosomal changes in cancer are acquired (somatic). Consistent with a multistep pathogenesis, most solid tumors exhibit multiple chromosomal abnormalities, and it is these changes that lead to an abnormal proliferation (clonal expansion) and, ultimately, to invasion of the surrounding tissues and metastasis to distant sites.12 The accumulation of chromosomal changes, which in most cases occur over a period of years, underlies both the process of tumorigenesis (the transition from normal cells to invasive cancer) and tumor progression (the transition to a metastatic, and often treatment-resistant, cancer). The karyotypes of many solid tumors (epithelial tumors in particular) are complex, and distinguishing “drives” (causative changes) from “passengers” (bystanders, victims of genomic instability) is often difficult. For example, in breast carcinomas, patients often exhibit heterogeneous karyotypes with multiple numerical and structural abnormalities (mostly unbalanced), and it is not possible to identify the primary aberration. Such complex karyotypes can be resolved and the breakpoints (balanced and unbalanced translocations) mapped by array painting, a method in which individual derivative chromosomes are purified using a fluorescence-activated cell sorter, amplified and hybridized to DNA microarrays. A study of 3breast cancer cell lines has shown that, contrary to the general view, a substantial proportion of translocation breakpoints are actually balanced.13
Chromosomal Changes In Solid Tumors: Clinical Significance
Sarcomas: Classification, Diagnosis, and Prognosis
Sarcomas comprise a heterogeneous group of mesenchymal neoplasms that arise in the bones and soft tissues. More than 100 distinct diagnostic subtypes, that range from benign, intermediate (locally aggressive), intermediate (rarely metastasizing), to malignant have been described.14 Such remarkable diversity always poses a challenge in the diagnosis, prognosis, and proper management of the disease. Over the past 2 decades our understanding of the pathobiology and genetics of the different subtypes of sarcomas has increased significantly. Cytogenetic analyses of over 2000 cases has revealed numerous chromosomal alterations that can be broadly classified into 4 categories9,14: (1) specific translocations that result in oncogenic fusion products or ectopic expression of proto-oncogenes (Figs. 1A–C, 2A–C; Table 1); (2) amplification of specific genomic loci/genes, such as amplification of MDM2/CDK4 in well differentiated liposarcomas, dedifferentiated liposarcomas, and intimal sarcomas. The amplified regions characteristically present as giant marker chromosomes and/or ring chromosomes (Fig. 3A–C); and (3) complex heterogeneous karyotypes with multiple structural and numerical abnormalities, such as in leiomyosarcoma, pleomorphic sarcomas, and giant cell tumors (Fig. 4A–B).


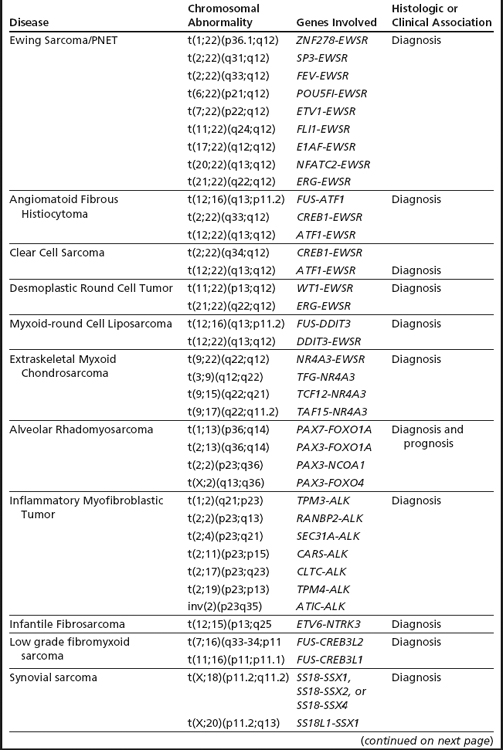
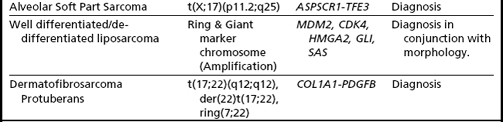


In addition to providing crucial clues to the genomic regions/genes involved in the development of sarcomas, these abnormalities serve as valuable markers in routine clinical practice in their classification, diagnosis, and prognosis (see Table 1). The classic example of classification/diagnosis is the small round blue cell tumors which includes the primitive neuroectodermal tumor (PNET) family, neuroblastoma, rhabdomyosarcoma, and lymphoblastic lymphoma of the bone. Although the majority of cases can be identified by morphology and immunohistochemistry, some of the undifferentiated cases continue to pose a diagnostic dilemma. The finding of t(11;22)(q24;q12)-EWS-FLI1 or its variants (EWS-ERG,EWS-ETV), observed in more than 95% of cases, confirms the diagnosis of PNET/Ewing’s sarcoma (see Fig. 1A). Similarly, the t(11;22)(p13;q12)/EWS-WT1 points to the diagnosis of small desmoplastic round cell tumors (see Fig. 1B), and the t(12;16)(q13;p11.2)/FUS-CHOP or its variants to the diagnosis of myxoid liposarcoma among the mixoid sarcomas (see Fig. 1C). The poorly differentiated or monophasic forms of synovial sarcoma can sometimes be challenging to distinguish from tumors such as malignant peripheral nerve sheath tumor, fibrosarcoma, primitive peripheral neuroectodermal tumor, or hemangiopericytoma. Similarly, the detection of t(X;18)(p11.2;q11.2)/SYT-SSX1 (see Fig. 2A) is required to make a diagnosis of synovial sarcoma. Other examples include the t(2;13)(q25;q14)/PAX3-FOXO1 (see Fig. 2B) and t(1;13)(p36;q14)/PAX7-FOXO1, found only in alveolar rhabdomyosarcoma. Histologically, rhabdomyosarcomas are divided into alveolar and embryonal subtypes, and it is important to distinguish the two. This is because the alveolar subtype carries an unfavorable prognosis and any attempt to improve longterm survival may require more intensive chemotherapy. The identification of specific translocations can also assist in the diagnosis of metastatic tumors whose origin cannot be established with certainty. Several other examples where chromosomal findings are clinically useful are listed in Table 1.
Renal Cell Carcinoma: Classification and Diagnosis
Renal cell carcinoma (RCC) is a heterogeneous group of diseases and conventional cytogenetic analyses have played an important role in the diagnosis and classification of this entity. Based on the pathologic and genetic features, the current WHO classification recognizes 2 broad categories, malignant and benign, and each has distinct histologic subtypes.15 The 3 main histologic subtypes are clear cell, papillary, and chromophobe, and respectively account for 80% to 90%, 10% to 15%, and 4% to 5% of all RCCs. The remaining histologic subtypes are rare. As shown in Table 2, each histologic subtype is associated with a unique combination of numerical gains and losses, deletions, and/or translocations. These alterations are extremely useful in the differential diagnosis of RCC.
Table 2 Recurrent chromosomal alterations in renal cell carcinoma (RCC) and neuroepithelial tumors (glioma)
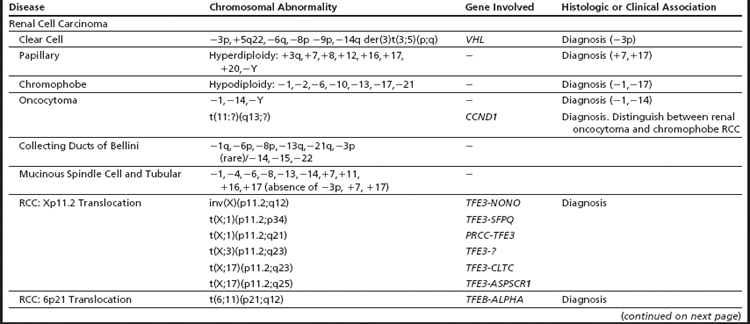
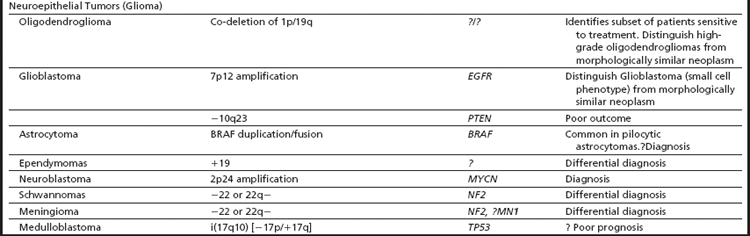
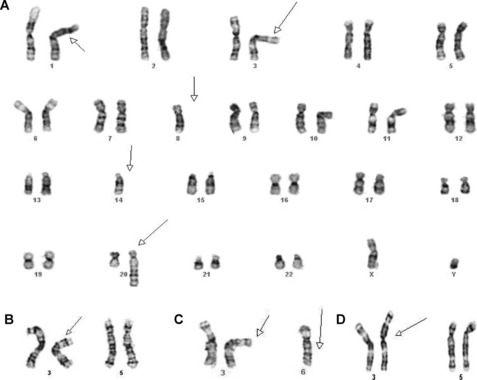
Cytogenetic and molecular studies have established that deletions of chromosome 3p (VHL and/or FHIT), resulting through monosomy 3 or various unbalanced aberrations, define the clear cell subtype (Fig. 5A–D). In up to 35% of the cases, an unbalanced translocation between chromosome 3p and 5q, resulting in the partial loss of 3p and gain of 5q, can be seen; it is reported to have a favorable outcome. Other frequent abnormalities include loss of 6q, 8p, 9p, and 14q, and these appear to be more common in advanced-stage disease. Deletions of 9p have also been associated with an unfavorable prognosis.16 The papillary subtype is defined by the simultaneous gains of chromosomes 7 and 17 with or without the loss of the Y chromosome. Additional numerical gains include chromosomes 3q, 8, 12, 16, and 20. The chromophobe subtype is defined by a hypodiploid karyotype with monosomies of chromosomes 1, 2, 6, 10, 13, 17, and 21. Loss of the Y chromosome may or may not be observed. The benign oncocytoma is heterogeneous with at least 2 distinct entities identified; one defined by a hypodiploid karyotype with combined loss of chromosomes Y, 1, and 14 and the other by promiscuous translocations of 11q13 resulting in overexpression of CCND1 (Cyclin D1) (Fig. 6A). Translocations of Xp11.2 involving the TFE3 gene (see Fig. 6B and Table 2), accounting for approximately 30% of pediatric and 15% of RCC patients under 45 years of age, identify patients that might benefit from targeted therapy. Several studies have also shown that the different histologic subtypes differ significantly in their clinical outcome and require different treatment approaches, including therapy. Thus, clinically, it is important to diagnose these tumors correctly.15,16

Over the last 2 decades, management of RCC has changed significantly. Due to the growing use of new and improved noninvasive abdominal imaging modalities, such as ultrasonography, CT, and MRI, more than 70% of RCC are detected, incidentally, as small renal masses in asymptomatic patients. These small asymptomatic masses are more frequently benign or indolent and, following histologic assessment on the biopsy specimen (core or fine-needle aspirate) and other diagnostic work-up, a proportion of patients (elderly and those with significant comorbidity) can enroll in an active-surveillance protocol and avoid the complications and costs of unnecessary surgery.17 One of the major concerns with core biopsies or fine-needle aspirate of renal masses is the risk of misdiagnosing or undergrading tumors as a result of overlapping histologic features or histologic heterogeneity. In this setting, assessment of histology-specific chromosomal alterations by FISH, along with histopathology, is proving to be useful in classification and treatment decision-making.18
Related posts:
 Cytogenetics
Cytogenetics
 Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
 Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
 In Situ Hybridization
In Situ Hybridization
 Myeloma: Current Perspectives
Myeloma: Current Perspectives
 Syndromes
Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


