Figure 17-1 Major events in vascular development Some of the critical signaling molecules and receptors are shown corresponding to the cells or processes in which they are known to play a role. Vascular progenitors are derived from vascular endothelial growth receptor-2 (VEGFR2/Flk-1)-positive cells in the lateral plate mesoderm. Hemogenic endothelial cells give rise to hematopoietic stem cells (HSCs) and vascular endothelial precursors (angioblasts). In the yolk sac, angioblasts align to generate a primary capillary plexus (vasculogenesis). Vessels in this plexus grow primarily by sprouting, which involves endothelial cell proliferation and migration (angiogenesis), and eventually connect to vessels in the embryo to form a closed vascular system. Vasculogenesis and angiogenesis are both highly dependent on VEGF, angiopoietins, and their receptors, along with many other signaling molecules (see figure and text). Maturation of the vascular system requires remodeling of the vascular network into large and small vessels, along with the recruitment of supporting mural cells (pericytes and smooth-muscle cells). Ang, Angiopoietin; Notch/Delta4, Notch receptor/Delta4 ligand; PC, pericyte; PDGF-β, platelet-derived growth factor β; PDGFR-β, PDGF receptor β; Robo/Slit, roundabout receptor/slit ligand; TGF-β, transforming growth factor-β; Tie2/Tek and Tie1, Tie family of endothelial receptor tyrosine kinases. (Adapted from Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671-674).
The vessels of the parallel lymphatic system collect and return interstitial fluids, particulates, and extravasated cells to the venous circulation. Lymphatic vessels differ from blood vessels in that lymphatic capillaries have internal membranous valves that prevent fluid backflow, and they are generally not surrounded by support cells. 9 Lymphatic ECs are derived from primitive veins and express and respond to a different spectrum of receptors and signaling molecules than ECs in blood vessels (Ref. 9, and see later discussion). The ability of cancer cells to invade lymphatics and collect in lymph nodes, complex organs involved in local immune surveillance, is an important indicator of tumor metastasis. It is likely that the lymphatic vessels at the periphery of a solid tumor are most directly involved in metastasis, as interstitial pressure within the tumor often leads to vessel collapse. 5,9 Recent evidence supports the idea that lymphatic ECs may secrete chemokines that attract tumor cells and may therefore participate more actively in metastasis than was previously recognized. 10
In adult humans and mice, there is little regular angiogenic activity, with the notable exception of the female reproductive system. Localized angiogenesis is, however, an important aspect of normal wound healing, and inflammatory cells including macrophages, neutrophils, and mast cells, as well as activated resident fibroblasts, are an important source of angiogenic modulators during wound repair. Recently, it was shown that macrophages directly bind angiogenic ECs and promote the formation of anastomoses between them. 11 On remodeling and fusion with the surrounding vasculature, these new vessels restore normal blood supply to the wounded area. These infiltrating stromal cells also represent an important component of many solid tumors, where they can produce angiogenic factors as part of what may be considered an aberrant wound healing response, leading to the idea that tumors represent “wounds that never heal.” 12 Genetic experiments using multiple murine cancer models have established that tumor-associated macrophages play a critical role in driving tumor angiogenesis and metastasis. 12
Rapid growth of any tissue (embryos, neoplasias, adipose tissue, regenerating liver, etc.) invariably requires a supply of oxygen, nutrients, and hormones and is typically accompanied by active angiogenesis. Consequently, angiogenesis can be seen as a genetically programmed, dynamic process that can be activated locally in response to stimulatory signals. The fact that most blood vessels in the adult body are quiescent has been proposed as an advantage of anti-angiogenic strategies, which typically target actively dividing ECs, as these drugs may be less generally toxic to quiescent ECs lining established vessels.
Tumor Vasculature
The blood vessels found in solid tumors are notable for being highly disorganized compared to those of normal organs and are characterized by tortuous and misshapen vessels that sometimes terminate in open-ended blood lakes 5,13 (Figure 17-3 ). These aberrant vessels are thought to result from dysregulated angiogenic signaling in the tumor bed, as a result of oncogene activation and tumor suppressor loss. Microscopic analysis of tumor vessels reveals disrupted junctions between tumor ECs and reduced or inconsistent coverage by pericytes, which helps explain the increased permeability characteristic of tumor vessels. 14 The origin of some tumor ECs is also controversial: In addition to ECs recruited through sprouting of preexisting vessels, growing evidence supports a role of circulating endothelial progenitor cells (EPCs) that either differentiate into endothelial-like cells or promote expansion of bona fide ECs (see Figure 17-2). The precise cellular origins and complexity of these cells remain controversial, and the degree to which murine EPCs actually contribute directly to the lining of new tumor vessels varies considerably, depending on the model used, genetic background, and other factors. 15,16 In addition, bone marrow–derived myeloid cells contribute to tumor angiogenesis; these cells have been reported to express a variety of cell surface markers, including those common to endothelial cells (Tie-2) and myeloid cells (CD11b, Gr-1), and may function by providing paracrine angiogenic signals. 17,18 It is interesting to note that genetic ablation of bone marrow–derived Tie-2 expressing monocytes (TEMs), in particular, has profound effects on tumor angiogenesis in mice (see Refs. 19 and 20, and references therein).
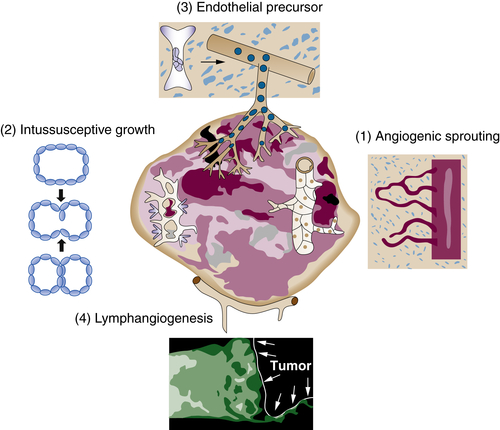
Figure 17-2 Cellular mechanisms of tumor angiogenesis Tumor vessels grow by multiple mechanisms, some of which are formally similar to those observed in normal vascular development: (1) budding of endothelial sprouts and formation of bridges (angiogenesis) and (2) insertion of interstitial tissue columns into the lumen of preexisting vessels (intussusception). In contrast to normal vascular development, the signaling events controlling these events are often highly disordered, resulting in chaotic vascular organization, uneven blood flow, and localized hypoxia. In addition, endothelial cell precursors home to tumors from the bone marrow or peripheral blood (3) where they can contribute, either directly or indirectly, to the endothelial lining of tumor vessels. Lymphatic vessels (4) around tumors drain interstitial fluid and also provide a gateway for metastasizing tumor cells. (Reproduced from Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249-257).
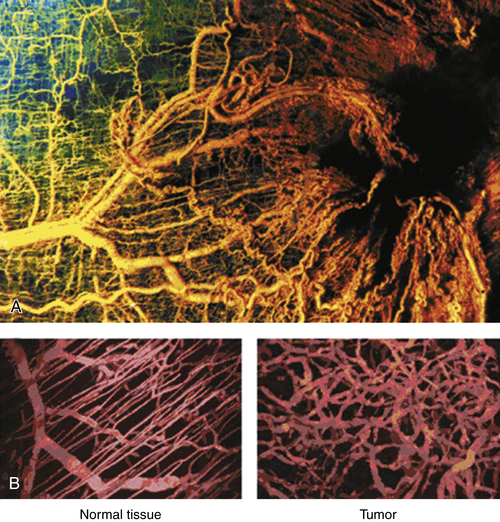
Figure 17-3 The highly disorganized nature of tumor vasculature can be visualized by generating a polymer cast before fixation (A) or using intravital imaging techniques that reveal functional vessels in live tissues (B). As opposed to the clearly ordered arrangement of vessels in normal tissue, the chaotic nature of tumor vessels reflects the disrupted balance of pro- and anti-angiogenic factors generated by tumor and stromal cells. (Reproduced from Weinberg RA. The Biology of Cancer. New York, NY: Garland Science; 2007:562).
Tumors often display sluggish, uneven, and highly variable patterns of blood flow, 13 as well as direct arteriole-venule shunts. 21 Tumor vessels also differ from normal vasculature in being exposed to an acidic microenvironment characterized by oxygen and nutrient deprivation. In rapidly growing tumors, aberrant angiogenic regulation and high interstitial pressure can produce regions of localized anoxia and/or ischemia. This typically results in pockets of necrosis surrounded by a penumbra of hypoxic but living cells. Severely hypoxic conditions are thought to protect tumor cells from radiation therapy, which depends on the generation of reactive oxygen intermediates to kill tumor cells. Moreover, hypoxic regions in tumors appear to select for highly malignant cancer cells. 22 In particular, hypoxia directly promotes angiogenic signaling in tumors, as discussed in more detail later. 23
The degree to which tumors generate vascular beds is often expressed as microvessel, or mean vessel, density (MVD), which can vary widely within a given tumor and between tumors of similar or different tissues. MVD is traditionally determined by staining tumor sections with antibodies raised against proteins expressed on ECs, including CD31 (PECAM), CD34, and von Willebrand factor. Clinical studies have demonstrated that MVD is a useful prognostic indicator for a wide array of cancers, including breast, prostate, non–small-cell lung, gastrointestinal, and even hematological tumors. 24 It is important to note, however, that not all tumor vessels are functional and that MVD may greatly exceed the basic metabolic requirements of a growing tumor. The striking functional heterogeneity of vessels within a tumor, and the ability of many cancer cells to withstand severe hypoxia, glucose deprivation, and tissue acidity, makes it difficult to assess the effects of angiogenesis-based therapies based solely on MVD. 24
Critical Signaling Factors—Targets for Therapy
Over the past 15 years, work from many laboratories has demonstrated that vascular development in normal tissues is under elaborate genetic and molecular control. Many of the signaling molecules that regulate normal developmental angiogenesis have also been shown to drive angiogenesis in cancer and other pathophysiological conditions, although their expression and function in tumors are often highly uncoordinated. A growing list of molecules has been shown to regulate different aspects of developmental and pathological angiogenesis. Primary among these is the family of vascular endothelial growth factors (VEGFs) that, along with their receptors, regulate endothelial cell proliferation, survival, and function. The vascular-specific angiopoietins and their receptor tyrosine kinases also play important roles in angiogenic remodeling. In addition, vascular development is regulated by signaling pathways familiar from other developmental processes, including fibroblast growth factors (in particular, basic or bFGF), transforming growth factor beta (TGF-β), Notch and its ligand Delta-like ligand 4 (Dll4), and platelet-derived growth factor (PDGF). In addition, a number of molecules originally implicated in controlling axon guidance, including the semaphorins, netrins, and Robo/slit, have been shown to contribute to vascular development. 7,25 Finally, the Notch pathway, along with the EphB4/ephrinB2 signaling system, has been shown to control specification of arteries and veins (see Refs. 7 and 25, and references therein). Our understanding of the mechanisms by which these genes and pathways regulate angiogenesis is based largely on genetic “knockout” experiments in mice, often confirmed by in vitro cell-based assays or in experimental tumors. How this complex array of signaling pathways is coordinated to regulate angiogenic events in normal organogenesis and disease is a focus of intensive research. The discovery of endogenous angiogenic inhibitors, including thrombospondin-1, endostatin, tumstatin, and others, provided strong support for the idea that angiogenesis regulated by the balance between pro- and anti-angiogenic factors. 26 In this section, we discuss the molecular biology and function of a small subset of pro-angiogenic and anti-angiogenic factors that show particular promise as targets for cancer therapies.
Pro-angiogenic Factors
VEGF
Vascular endothelial growth factor (also known as VEGF-A) is among the most potent angiogenic factors described and stimulates EC proliferation, survival, chemotaxis, and vessel permeability. VEGF belongs to a family of structurally related growth factors that includes placental growth factor (PlGF), VEGF-B, VEGF-C, and VEGF-D. VEGF is a homodimeric glycoprotein of 45 kDa and is expressed in four different molecular-weight forms—VEGF-121, VEGF-165, VEGF-189, and VEGF-206—produced by differential mRNA splicing. VEGF-121 is diffusible, whereas the other forms bind to heparin and heparin proteoglycans in the extracellular matrix (ECM) and on cell surfaces. These bound forms are released through the action of proteases, including plasmin and matrix metalloproteases (MMPs), which are produced by tumor cells and/or by activated stromal cells. Interestingly, VEGF was first identified as vascular permeability factor (VPF) based on its ability to increase the leakage of fluid and plasma proteins from blood vessels (see Refs. 1, 5, 14, and 27 for details). These leaked proteins provide an ECM for migrating ECs, and their release into interstitial spaces represents an early step in angiogenesis. The central importance of VEGF in regulating angiogenesis became clear through genetic targeting experiments in mice. Loss of only one Vegf allele resulted in lethality at embryonic day 9.5 (E9.5), characterized by a reduction in ECs and abnormal vessel morphology. 28,29 Embryos lacking both Vegf alleles died even earlier (E8.5) and displayed a complete absence of the dorsal aorta and other vascular structures.
VEGF mediates its effects by binding its cognate receptor tyrosine kinases, VEGFR1 (also called Flt-1) and VEGFR-2 (also called Flk-1 or KDR). Binding of VEGF to VEGFR-2/Flk-1 triggers receptor autophosphorylation and robustly activates several downstream signaling pathways (including phosphoinositide 3-kinase [PI3K], Src, and protein kinase C [PKC]), leading to rapid and profound effects on EC proliferation, survival, migration and gene expression. 7,30 Genetic ablation of Flk-1 in mice caused embryonic lethality at E8.5 that correlated with a loss of normal vascular structures and hematopoietic cells, consistent with the bipotential fate of hemogenic endothelial cells. 8,31 Subsequent studies have confirmed the importance of VEGF and VEGFR-2/Flk-1 in hematopoietic development (Ref. 7 and references therein). Although VEGFR-1/Flt-1 also binds VEGF, its major angiogenic function may be to modulate the amount of VEGF available to bind to VEGFR-2/Flk-1. 14 Deletion of the gene encoding murine VEGFR-1/Flt-1 resulted in embryonic lethality; however, this lethality was rescued by transgenic expression of a truncated VEGFR-1/Flt-1 protein that lacked its cytoplasmic signaling domain. Although these results argue strongly that VEGFR-1/Flt-1 acts as a nonsignaling sink for free VEGF, subsequent studies indicate that it can, in fact, modulate pathophysiological angiogenesis, possibly by intermolecular phosphorylation of VEGFR-2/Flk-1. 9 Neuropilins 1 and 2 can also act as a sink for VEGF and appear to function, at least in part, by presenting VEGF to VEGFR-2/Flk-1 or by modulating its effective free concentration. 32
The central role of VEGF signaling in tumor angiogenesis has been clearly demonstrated in a wide variety of experimental models, including VEGF overexpression in tumor or host cells, treatment with recombinant VEGF, increased VEGF expression in response to oncogene activation, or inhibition by antisense VEGF oligonucleotides or anti-VEGF antibodies. 1,25 Furthermore, many oncoproteins (including KRAS, HER2, FOS, and TRKB), tumor suppressor proteins (including pVHL and p53), and growth factors (including PDGF, bFGF, and TGF-β) regulate angiogenesis, partly by inducing the expression of VEGF either directly or indirectly. 25
The von Hippel–Lindau (pVHL) tumor suppressor is a particularly interesting case in point. Patients with VHL disease, a hereditary cancer syndrome, develop a variety of tumor types including highly vascularized renal clear cell carcinomas, cerebral hemangioblastomas, and retinal hemangiomas. The pVHL protein functions as an E3 ubiquitin ligase that targets the hypoxia inducible factor (HIF) subunits HIF-1α and HIF-2α for oxygen-dependent degradation via the 26S proteasome. 33 HIF-1α and HIF-2α play a predominant role in hypoxic responses, 34 and their activity is controlled in a similar oxygen-dependent fashion. Both proteins regulate the expression of target genes that mediate adaptive responses to hypoxic stress, including those encoding VEGF and many other angiogenic factors 23,35 (see Figures 17-3 and 17-4 ). When pVHL expression or function is lost, cells can no longer degrade the HIF-α subunits under conditions of abundant oxygen, leading to constitutive expression of VEGF and other HIF target genes, thereby promoting tumor angiogenesis (Figure 17-5 ). Both HIF-α subunits are often overexpressed in cancer cells as a consequence of oncogene activation, tumor suppressor loss, or tumor hypoxia. The close spatial overlap between HIF-α protein accumulation and VEGF expression in hypoxic tumor cells is a further indication that HIF-dependent VEGF expression is an important aspect of tumor angiogenesis (see Figure 17-3).
Both HIF-1α and HIF-2α bind to the Vegf gene promoter and can activate VEGF expression independently; hence, deletion of either subunit has relatively subtle effects on embryonic VEGF expression, despite the fact that both mutations are embryonically lethal. 23,36–38 Targeted deletion of the common binding partner (HIF-1β or ARNT), however, resulted in early embryonic lethality with substantial loss of VEGF expression 39 associated with fundamental defects in angiogenesis. 40 The close link between the HIFs and VEGF expression in tumors has prompted the design of specific HIF inhibitors, partly to limit expression of VEGF and other hypoxically induced angiogenic factors in cancer and other diseases. 41,42
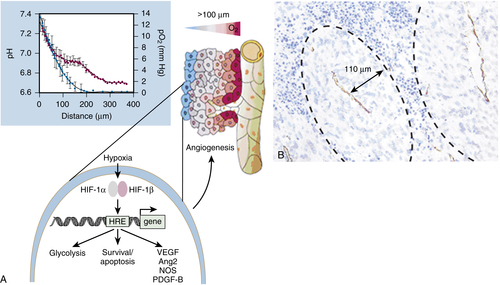
Figure 17-4 (A) Because of the irregular pattern and organization of the tumor vasculature, some cells in tumors are located more than 100 m (the diffusion limit for oxygen) away from blood vessels and become hypoxic (red-to-blue gradient indicates progressive hypoxia). Tumor cells survive fluctuations in oxygen tensions, in part because clones are selected in hypoxic tumors that switch to a pro-angiogenic phenotype. HIFs increase transcription of several angiogenic genes (for example, genes encoding VEGF, PDGF-BB, and nitric oxide synthase [NOS]). HIFs also affect cellular survival/apoptosis pathways. Inset: Relationship between the distance of tumor cells from nearby vessels and their degree of hypoxia (blue symbols) and acidosis (red symbols). (Reproduced from Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249-257). (B) Section of rat prostatic carcinoma in which vessels were identified by CD31 immunostaining. A “cuff” of viable cells surrounds each capillary, beyond which regions of necrosis are evident. (Reproduced from Hlatky L, Hahnfeldt P, Folkman J. Clinical application of antiangiogenic therapy: microvessel density, what it does and doesn’t tell us. J Natl Cancer Inst. 2002;94:883-893).
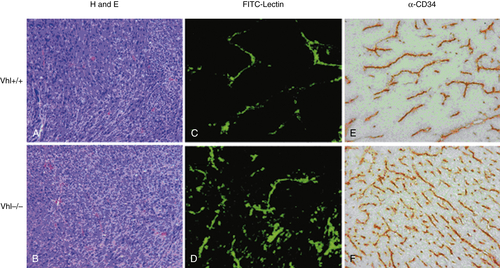
Figure 17-5 Loss of the pVHL tumor suppressor increases tumor angiogenesis Fibrosarcomas were generated subcutaneously in immunocompromised mice by injecting Ras-transformed fibroblasts derived from wild-type (Vhl +/+ ) or pVHL-deficient (Vhl –/– ) mice. Tumor sections reveal that loss of pVHL, and consequent constitutive HIF activation, correlated with increased tumor angiogenesis. Tumor vessels were labeled with either FITC-lectin (C, D) or CD34 antibodies (E, F). H and E, Hematoxylin and eosin (A, B). (Courtesy F. Mack and M. C. Simon).
Each of the several VEGF homologs in mammals (VEGF-B, VEGF-C, VEGF-D, and PlGF) has distinct influences on angiogenesis and binds to one or more of the family of VEGF receptors. VEGF-C and VEGF-D regulate lymphangiogenesis through their effects on VEGFR-3/Flt-3, which is expressed on lymphatic ECs. 9 PlGF binds to both VEGFR-1/Flt-1 and the neuropilins, displacing VEGF and thereby making it available for binding to VEGFR-2. 32 Some data, however, suggest that heterodimers of PlGF and VEGF may be more potent in some contexts than the more typical VEGF homodimer. 14 Much work remains to be done to tease apart the unique and overlapping functions of the various VEGF homologs and their receptors.
bFGF
Basic FGF (bFGF or FGF2) is one of more than 20 known fibroblast growth factors that mediate a large number of developmental and homeostatic functions in different tissues. bFGF was identified biochemically in a search for angiogenic molecules released by tumor cells. When added to tissues exogenously or overexpressed in transplanted tumor cells, bFGF has potent angiogenic properties. 43 Like VEGF, bFGF binds to extracellular heparan sulfate proteoglycans and activates cognate receptor tyrosine kinases. Interestingly, loss-of-function studies have failed to reveal an inherent role of bFGF in embryonic angiogenesis, although this may be due to functional complementation by other FGF family members. As many of the angiogenic properties of bFGF appear to require VEGF function, however, one important role of bFGF in tumor angiogenesis may be to induce VEGF expression. 43 The situation is almost certainly more complex, because VEGF and bFGF act synergistically in some contexts but clearly have independent effects on ECs in others. The emerging picture suggests that bFGF and many other angiogenic factors act as general growth and survival factors for ECs partly by regulating VEGF expression, whereas VEGF itself may preferentially stimulate many of the cellular processes that lead to new vessel formation.
Angiopoietins/Tie Receptors
In addition to VEGF and FGF receptors, ECs express the Tie1 and Tie2/Tek receptor tyrosine kinases. Genetic ablation of either Tie1 or Tie2 in mice produced embryos in which vasculogenesis was intact, but subsequent angiogenic remodeling was inhibited. Soluble forms of these receptors were used to identify endogenous ligands, called angiopoietins (Ang1-4) (reviewed in Ref. 44). Deletion of Ang1 produced a phenotype similar to loss of Tie2, supporting a role for Ang1 as an important activator of Tie2 signaling. Interestingly, Ang2 also binds to Tie2 with high affinity but does not stimulate Tie2 tyrosine phosphorylation or downstream signaling. Transgenic overexpression of Ang2 produced a phenotype similar to that associated with loss of Ang1 or Tie2, suggesting that Ang2 may be a naturally occurring inhibitor of Ang1 signaling. The role of Ang2 became clearer when it was found to be induced in concert with VEGF at sites of vascular remodeling. Several studies have suggested a model in which Ang2 interferes with the stabilizing effects of Ang1 (such as increased pericyte and smooth muscle recruitment), thereby allowing VEGF to stimulate EC division and migration more efficiently. The roles of Ang3 and Ang4 are less clear, and a cognate ligand for Tie1 has not yet been identified, 44 although Tie1 may act primarily as a repressor of Tie2 signaling. 45
PDGF
Maturation and maintenance of the vascular system require the establishment of a close functional relationship between ECs and pericytes (PCs). ECs undergoing active division and morphogenesis express PDGF-B, and PCs express the corresponding receptor PDGFRβ. Genetic ablation of either ligand or receptor in mice disrupts PC recruitment, resulting in leaky, malformed blood vessels and increased EC apoptosis. 46 Bergers and colleagues identified a population of c-Kit+, Sca-1+ bone marrow–derived progenitor cells that are recruited to perivascular sites in tumors, where they differentiate into PCs and stabilize the tumor vessels in a PDGFRβ-dependent manner. 47 Overexpression of PDGF promoted recruitment of PCs and tumor vessel stabilization, whereas inhibition of PDGF signaling reduced PC recruitment with a concomitant increase in EC apoptosis. 25 Consequently, a combination of therapies that target both tumor ECs and PCs may prove to be a particularly effective approach. 25,48
Anti-angiogenic Factors
In his landmark 1971 paper, Judah Folkman not only proposed that tumor growth depends on angiogenesis, but also suggested that endogenous angiogenic inhibitors could be identified and used therapeutically. 4 Intensive efforts over the subsequent three decades have led to the identification of more than 30 endogenous inhibitors whose application can block angiogenesis in a variety of assays and genetic models. 49,50 These naturally occurring compounds include proteolytic cleavage products of extracellular matrix proteins (thrombospondin, endostatin, tumstatin), the protease plasminogen (angiostatin), and clotting factors (cleaved antithrombin III and prothrombin kringle-2), as well as immune modulators such as interferons and interleukins. 49 The specific function of each of these compounds in tumor angiogenesis, and their possible utility as therapies for cancer treatment, continues to be an area of active investigation.
Thrombospondin 1 (TSP-1)
Initially identified as an extracellular glycoprotein with cell-adhesive properties, TSP-1 binds to integrin and nonintegrin cellular receptors, cytokines, growth factors, and extracellular proteases. TSP-1 is thought to act as a molecular scaffold that facilitates interactions between proteins that regulate cell morphology, signaling, and adhesion, possibly by promoting receptor clustering. 51 In 1990, Bouck, Polverini, and colleagues described the strong anti-angiogenic activity of a TSP-1 proteolytic fragment. 52 Targeted deletion of TSP-1 in mice increased tumor angiogenesis and growth, and subsequent reports confirm the inability of TSP-1 mutant mice to mount a normal angiogenic response in other assays. 53 The Tsp-1 gene has been shown to be a direct target of the p53 tumor suppressor, and TSP-1 expression has been inversely correlated with the progression of carcinomas and melanoma in humans. 49 The molecular mechanisms by which TSP-1 blocks angiogenesis are likely to be complex, but may include integrin inhibition, interference with VEGF and bFGF signaling, and/or induced expression of the pro-apoptotic FasL protein on ECs. 49 The identification of the gene encoding TSP-1 as a direct p53 target suggests yet another mechanism whereby p53 inactivation can promote tumor progression.
Endostatin and Tumstatin
Both endostatin and tumstatin are proteolytic cleavage fragments derived from collagen molecules. Endostatin was initially purified from a murine hemangioendothelioma cell line and identified as a 20-kDa carboxy-terminal fragment of type XVIII collagen. Recombinant endostatin has multiple anti-angiogenic properties, including the ability to interfere with VEGF and bFGF signaling, inhibit EC motility, and induce EC cell cycle arrest and apoptosis. 49 Endostatin appears to mediate these pleiotropic effects by binding EC integrins, including α5β1, αVβ3, and αVβ1. Tumstatin consists of a 28-kDa fragment of the α3 chain of type IV collagen, promotes EC apoptosis, and suppresses the growth of various human tumor cells in xenograft experiments. Similar to endostatin, tumstatin binds to integrins and thereby inhibits activation of downstream signaling pathways. 49,50 Despite their similarities, endostatin and tumstatin peptides share little sequence identity and can clearly mediate independent functions: For example, endostatin inhibits EC migration with little effect on VEGF-induced proliferation, whereas tumstatin inhibits EC proliferation without significantly affecting migration.
It is interesting to note that many endogenous angiogenesis inhibitors are generated by proteolytic degradation of ECM proteins, or from proteins involved in blood clotting, and that many bind directly to integrin receptors. Growing evidence supports the notion that these compounds play an important role in fine-tuning the angiogenic response that accompanies thrombosis and tissue repair. 54 The production of these endogenous angiogenesis inhibitors may also help explain tumor dormancy, as first proposed by Folkman in 1971. Control of local angiogenic activity in a tumor is thought to be determined by the balance of pro-angiogenic factors (VEGF, angiopoietin 1, bFGF, etc.) and angiogenesis inhibitors (TSP-1, endostatin, tumstatin, etc.). Consequently, it may take months or years to generate the proper genetic and physiological conditions necessary to tip the balance to favor active blood vessel development and tumor growth—an event called the angiogenic switch. 55
Multiple lines of evidence from preclinical models, as well as patient data, support the role of an angiogenic switch in regulating tumor growth. For example, several murine genetic models have shown that tumors generated by transgenic expression of oncogenes initially remain small, with tumor cell proliferation largely offset by apoptosis. After a period of relative stasis, the tumors begin to show evidence of increased vascularity, after which they grow rapidly. 55 The synthesis of angiogenesis inhibitors by a primary tumor may also keep distant metastases from progressing, as removal of a large primary tumor often correlates with the rapid outgrowth of previously unidentified metastatic tumors in patients. 4,50 It has also been suggested that bone marrow–derived EPCs may play an important role in controlling this event and may regulate a critical step in the progression of micrometastases into macrometastases. 16 Collectively, a growing body of data suggests that manipulating the angiogenic switch can control tumor growth; this has prompted the development of clinically relevant angiogenesis inhibitors as cancer therapies.
Targeting Tumor Angiogenesis in Patients
Over the past 15 years, a large number of clinical trials have been conducted to test the efficacy of anti-angiogenic compounds in cancer therapy. Early results, however, were mixed: For example, early trials of endostatin and other compounds that showed promise in preclinical models yielded disappointing results in the clinic, 56,57 although exceptions included a trial showing positive effects of angiostatin in treating non–small-cell lung cancer when combined with cytotoxic chemotherapy. 58 Some possible reasons for the apparent discrepancies between dramatic results in preclinical models and actual patient responses are discussed next. Despite this somewhat rocky beginning, a number of targeted therapies have shown clinical benefit and have attained approval from the U.S. FDA and the EMA (European Medicines Agency) for treating cancer. In particular, the prominent role of VEGF in tumor angiogenesis made it an obvious therapeutic target, and multiple drugs have been developed that either sequester free VEGF (reducing its effective concentration) or block VEGFR-dependent signaling. The large number of preclinical and clinical studies on drugs directed against VEGF signaling provides an instructive paradigm for the potential successes and shortcomings of other anti-angiogenic strategies to treat cancer.
VEGF-Based Therapies
In 1993, Ferrara and colleagues reported that a murine anti-human VEGF monoclonal antibody could inhibit the growth of different human tumor cell lines in immunocompromised mice, although the antibody had no effect on tumor cell proliferation in vitro. 59 Subsequent analysis revealed that the antibody blocked angiogenic activity in these xenografts, and this led to the development of a humanized version of the antibody, called bevacizumab or Avastin, for human clinical trials. In 2003, results from two clinical trials of bevacizumab function generated tremendous excitement in the field. In one phase III trial, patients with advanced metastatic colorectal cancer were treated with bevacizumab in conjunction with cytotoxic chemotherapy 60 and displayed an average increase in overall survival (OS) of approximately 4 months (from 16 to 20). Although this response seems modest, it was the first indication that specific targeting of VEGF in highly metastatic human cancer could have a survival benefit. In a separate phase II trial, patients with metastatic renal cancer showed a significant, dose-dependent increase in progression-free survival (PFS) when treated with bevacizumab compared to placebo. 61 Interestingly, an Fab fragment of bevacizumab (marketed as Lucentis) has shown great success in treating patients with the angiogenic, or “wet,” form of age-related macular degeneration. 62
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


