Fig. 2.1
Detailed genetic composition on human MHC (HLA) including type 1 diabetes risk
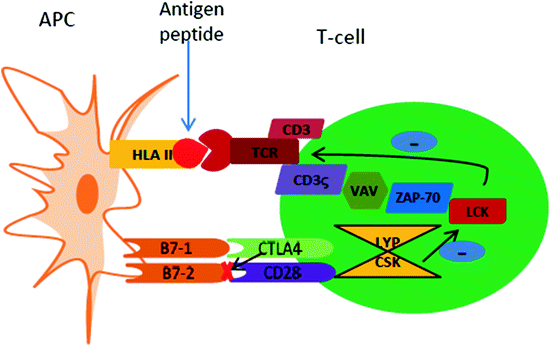
Fig. 2.2
Communication and interaction pathways between an APC and T-cell. APC presents a processed antigen peptide via HLA II on to the TCR/CD3 complex present on the T-cell surface. For the full activation of the T-cell, a second signal is required and this signal is generated via costimulatory molecular interaction between CD28 on the T-cell surface and B7-1 (CD80) and B7-2 (CD86) on the APC surface. Additional signaling messengers are present in the cell cytosol with positive regulatory tyrosine kinases such as LCK, CSK, VAV, ZAP-70 and CD3-ζ. These tyrosine kinases are inhibited by the LYP/CSK complex. CTLA-4 molecule is an inhibitory molecule, which competes with CD28 for occupying the B7-1 or B7-2 pockets; this inhibits the required secondary or costimulatory signal for T-cell activation leading to quiescent or apoptotic T-cells (Illustration modified from Wiebolt et al. 2010)
Table 2.1
Role of non-HLA genes in susceptibility
Non-HLA genes and chromosome location | PTPN22 (1p13.3-13.1) | INS (11p15.5) | CTLA-4 (2q33.2) | IL2RA/CD25 (10p15.1) |
|---|---|---|---|---|
Role in T1D | Encodes LYP; a dominant negative antigen-TCR regulator that acts via dephosphorylation and inactivation of T-cell | Encodes pre-proinsulin peptide (insulin precursor) | Surface molecule present on T-cells; negative regulator of TCR activation; blocks B7-1 and B7-2 (also called CD80 & CD86) (see Fig. 2.2) | Encodes α-chain of IL-2 receptor complex (also called CD25) essential for self-tolerance of Treg cells |
Major findings/Mutations | SNP at base pair 1858 from cysteine to thymine (1858C → 1858T) resulted in amino acid change from arginine to tryptophan (R620 W) Mutation found higher in T1D population compared to healthy controls | VNTR regulates INS expression; VNTR classification: • Class I: 2–5 fold increase in T1D risk (26–63 repeats/ ̴ 0.57kbp) • Class II: Most rare allele (80–85 repeats/ ̴ 1.64kbp) • Class III: Increased expression in thymus and decreased in pancreas and results in efficient thymus selection and protects against T1D (141–209 repeats/ ̴ 2.4kbp) | Threonine to alanine (A49G) substitution lead to abnormal post-translational glycosylation and decreased CTLA-4 surface expression and reduced Treg cells. Cytosine to thymidine (C318T) leads to increased CTLA-4 cell surface expression and hence imparts protection against T1D | Different IL-2R SNP have been associated with high risk of T1D such as rs41295061 and ss52580101; rs12722495 found to alter Treg function |
References |
2.1.1.2 Non-Human Leucocyte Antigen (Non-HLA Genes)
HLA genes account for some 40–50 % of genetic predisposition, suggesting that supplementary risk factors also play a near equivalent role in T1D progression. Efforts to identify such non-HLA genes have been possible, most notably through Genome-wide association studies (GWAS) and Gene-linkage studies (Davies et al. 1994; Hirschhorn and Daly 2005). GWAS is an approach which involves scanning markers, which can rapidly scan across the genome of an individual to find genetic variations which are associated with a particular disease (National Human Genome Research Institute 2014). Genetic linkage studies are used to identify regions of the genome that contain genes that predispose an individual to disease. Linkage analysis is used to map genetic loci by use of observations of related individuals (Dawn and Barrett 2005). Analysis of the available data suggests that non-HLA genes have relatively smaller yet significant effect on individual risk. A list of non-HLA risk associated genes with their chromosomal location, role in T1D and mutations are highlighted in Table 2.1.
Targeting the protective genes is considered a valuable approach to T1D therapy. However, there are various barriers before their successful translational potential can be harnessed. For example, there is a need for meticulous mechanistic studies to differentiate the dual role of several genes such as PTPN22, which is purported to protect against tuberculosis (TB), while predisposing patients to both T1D and rheumatoid arthritis (RA). Additional barriers include inadequate number of study sample sets and mixed ethnic groups that further add to the complexities and our understanding of dominant functionalities in T1D.
2.1.2 Environmental Triggers
The unexpected peak in the number of T1D patients across the globe represent strong evidence that the increase cannot be due to genetic susceptibility alone, as genetic changes present over a significantly longer period of time. This suggests involvement of factors, distinct from genetics such as environmental triggers or precipitating agents in the etiology of T1D (Åkerblom and Knip 1998). The hypothesis is supported by confounding geographic variation observed within the European population with a mere 3.2 cases/100,000/year reported in Macedonia, as opposed to a staggering 40.9 cases/100,000/year in Finland (Karvonen 2006). Moreover, migratory studies have shown higher incidence of T1D in individuals, who relocated from regions with low incidence to those with higher incidence (Tull et al. 1992). With this in mind, we next discuss the epidemiological data from various studies, which convince us of the association of several putative environmental triggers such as viruses, cow’s milk, vitamin D, and toxins (N-nitroso compounds) with T1D (Knip et al. 2005).
2.1.2.1 Viruses
The very first idea that manifestation of viral infections is linked to T1D etiology dates back to 1920s, where T1D onset was observed to follow a seasonal pattern (Adams 1926). Since then various viral candidates have been investigated for their role in the precipitation of the disease, such as enteroviruses (EVs) most notably coxsackie B virus (CBV), rubella, mumps, rotavirus, cytomegalovirus, and Epstein Barr virus (EBV). However, the evidences of association of viruses with T1D hitherto, have either been weak, irreproducible, and sometimes even been rejected by the scientific world due to the contradictions in the reports (Coppieters et al. 2012). Conversely, there are also a range of viruses which demonstrate a protective effect against autoimmune development, although they are deemed outside the remit of this book.
Enteroviruses (EVs) are among the most robustly reported infectious agents having a close association with T1D. EVs belong to the picornavirus family, these being small, naked, icosahedral, single stranded RNA-containing viruses. The first EV-T1D correlation was reported in 1969 (Gamble et al. 1969) while Oikarinen et al. only recently succeeded in isolating EVs from pancreatic biopsies of T1D patients (Oikarinen et al. 2008). Approximately 75 % (n = 12) of T1D patients versus 10 % (n = 10) in control subjects were found to have EV in their gut mucosa. Apart from only a handful of investigations reporting infants with increased HLA susceptibility toward T1D, several other studies present a contradictory view of the EV-T1D link. For instance, a prospective study conducted in Finland (DIPP) versus Colorado, USA (Diabetes Autoimmunity Study in the Young, DAISY) and Germany delivered contrasting verdicts with no report of increased T1D risk due to EV infection (Füchtenbusch et al. 2001; Graves et al. 2003; Salminen et al. 2003). However, it is difficult to confirm, with any level of precision whether the reported events are a result of differences in the methodology employed, or if in fact there is an actual difference, within the screened populations. Further, these studies lead to the corollary that there is a need to first identify whether the viral infections are acute or persistent in nature (Tauriainen et al. 2011) as well as assessing if the relationship found in some cases is really a causal effect or just a secondary event in patients already presenting clinical diabetes. It is interesting to note that parallel studies on another strain of coxsackie viruses, i.e. CBV3, resulted in delayed onset of diabetes due to expansion of CD4+CD25+ Treg (regulatory T-cells) cells (Viskari et al. 2000, 2005). In other words, time of infection and inoculation dose have a significant role to play in the development of T1D.
Two different mechanisms have been proposed to explain the causative role of viruses in T1D. First, “molecular mimicry” which is described by resemblance of the P2-C protein sequence of the coxsackie virus with that of glutamic acid decarboxylase 65 (GAD65) epitope, a major antigen determinant of T1D (Hou et al. 1994). It was observed that T-cells from patients with higher risk of T1D, which responded to GAD65 determinants, also responded to the P2-C peptide of the virus (Helgason and Jonasson 1981; Schloot et al. 2001). However, autoreactive human T-cell clones that are specific for GAD65 epitope failed to proliferate poststimulation with the viral epitope (Schloot et al. 2001). Further studies in rat insulin-promoter lymphocytic choriomeningitis virus (RIP-LCMV) mice suggested that in order to initiate viral infection, it is necessary to have a complete homology between viral and β-cell antigens, since a mere single amino acid change flanking a cytotoxic T-lymphocyte (CTL) epitope was found to interfere with the development of T1D (Fujinami et al. 2006; Knip et al. 2010). The second hypothesized mechanism involved in the viral etiology of T1D is “bystander activation”. It is proposed that this bystander mechanism works via a TCR-independent mechanism (Jun and Yoon 2003). The substitute mechanisms contain activation through soluble factors or membrane-bound molecules that bind to receptors other than TCR. Several other mechanisms include the alteration of the host immune system followed by viral infection.
These proposed mechanisms are far from being able to explain all the observations. To further confuse matters there have been additional findings suggesting an inverse relationship between EVs and T1D incidence. In Finnish and Swedish studies, a decrease in the frequency of EV infection has been reflected by increased incidence of T1D compared to other countries, such as Estonia and Russia; this was later explained by the “hygiene hypothesis” (Viskari et al. 2005). Conversely, it has also been observed that childhood immunization against certain diseases of viral origin provide a protective effect against T1D (Classen and Classen 1999, 2002).
2.1.2.2 Dietary Factors
Viral infection and genetic susceptibility are not the only culprits triggering T1D. Rather, recent changes in early life diet and feeding habits implicate several dietary components as additional triggers driving autoimmune T1D. The following section discusses some of the known dietary factors linked to T1D etiology.
2.1.2.3 Early Dietary Habits—Cow’s Milk and Gluten
The early exposure to cow’s milk and the nutritional benefits of breast milk have been extensively investigated with outcomes having been reported. Indeed, a study amongst the Scandinavian population, showed an inverse relationship between breastfeeding and T1D incidence (Borch-Johnsen et al. 1984), while a Finnish study reported that short-term breastfeeding followed by early introduction of a cow’s milk-based infant formula predisposed young children to progressive signs of β-cell autoimmunity, rendering these children genetically susceptible to T1D (Kimpimäki et al. 2001).
Other investigation focused on the role of cow’s milk with supplementation from various dietary components, such as early introduction of solids and gluten containing diets. Gluten is a protein principally found in wheat and it can be further divided into two subgroups of protein, the gliadins and glutenins. Anecdotal reports claim a low incidence of T1D in rodents with diets containing low levels of protein such that inclusion of gliadin by as little as 1 % by weight, of individuals total food consumption showed T1D incidence of about 35 % compared to the 15 % incidence in control group fed on a semi-synthetic diet (Elliott and Martin 1984). Separately, two prospective cohort-based studies arrived at a conclusion that a “critical period of exposure” to cereals is associated with the progression of T1D. Norris et al. found that children exposed to cereals from 0 to 4 months of age and those exposed at an age of 7 months or older have higher risk of developing autoimmunity with hazard ratio (i.e., hazard in the exposed group/hazard in the unexposed group) (HR) of 4.32 and 5.36, respectively (Norris et al. 2005). A German study reported similar results with a five-fold (HR of 4.0) higher risk of T1D development in children exposed to a gluten containing diet before age of 3 months compared to those exposed after the age of 6 months (Ziegler et al. 2003). The risk associated with the early exposure may be due to anomalous immune response to cereal antigens in an immature gut system of susceptible individuals, while the late exposure risk in older children could be attributed to the consumption of larger portions of cereals.
The inferences from the above studies are limited by numerous methodological incongruences, such that discrepancies in the reported outcomes may well be attributed to differences in the study population or to the variation in the feeding practices across the countries where the studies were conducted. Hence it is imperative to address this issue systematically, as attempted in the ‘Trial to Reduce Insulin-dependent diabetes mellitus (IDDM) in the Genetically at Risk (TRIGR).’ In a recent, albeit limited scope pilot study, and the first of this kind in humans, a link suggesting that it is possible to manipulate spontaneous β-cell autoimmunity by nutritional intervention during infancy was reported (Åkerblom et al. 2005). TRIGR, an international group conducting collaborative clinical trials at 77 clinical centers in 15 different countries across 3 continents, began addressing the hypothesis of whether weaning with a hydrolysate protein-containing diet decreased T1D incidence in a genetically susceptible population, compared to that of a cow’s milk protein diet (Åkerblom 2011; Knip et al. 2014). Successful collaborative data from the trial is expected to be major stepping stone toward primary prevention and intervention strategies of T1D.
Related posts:
 Predictors and Pathogenesis of Type 1 Diabetes
Predictors and Pathogenesis of Type 1 Diabetes
 Introduction to Diabetes and Type 1 Diabetes
Introduction to Diabetes and Type 1 Diabetes
 Type 1 Diabetes: Past, Present, and Future Therapies
Type 1 Diabetes: Past, Present, and Future Therapies
 Female reproduction: VI Contraception
Female reproduction: VI Contraception
 Psychological Stress, Immunity, and the Effects on Indigenous Microflora
Psychological Stress, Immunity, and the Effects on Indigenous Microflora
 Cardiovascular and Metabolic Syndrome Impact on Uristatin
Cardiovascular and Metabolic Syndrome Impact on Uristatin
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


