Several host and clinical factors are associated with an increased risk of SMNs after HCT. These include age at HCT, exposure to chemotherapy and radiation prior to HCT, use of total-body irradiation (TBI) and high-dose chemotherapy for myeloablation, infection with viruses such as Epstein-Barr virus (EBV) and hepatitis B and C viruses, type of transplantation (autologous versus allogeneic), and immunodeficiency after HCT aggravated by the use of immunosuppressive drugs for prophylaxis and treatment of graft-versus-host disease (GVHD).1
The heterogeneous nature of SMNs, with differing clinicopathologic characteristics, precludes assessment of risk factors in aggregate. It has become conventional practice to classify SMNs into three distinct groups12: (1) myelodysplasia (MDS) and acute myeloid leukemia (AML), (2) lymphoma, including lymphoproliferative disorders, and (3) solid tumors. Although secondary leukemia and lymphoma develop early in the posttransplantation period, secondary solid tumors have a longer latency. In this chapter, the three broad categories of SMNs seen among patients undergoing HCT are discussed, with emphasis on clinical presentation, magnitude of risk and associated risk factors, current insights into pathogenesis, and treatment options and outcome of patients with these malignancies. We will conclude the chapter with a brief summary regarding strategies for early detection of SMNs.
MYELODYSPLASIA AND ACUTE MYELOID LEUKEMIA
Therapy-related MDS (t-MDS) and therapy-related AML have emerged as the major cause of nonrelapse mortality in patients undergoing autologous HCT for Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL).4,13 The risk of t-MDS/AML approaches 9% after autologous HCT for lymphoma with a median latency of 12 to 24 months after HCT (range, 4 months to 6 years).4
Clinicopathologic Syndromes
Two types of t-MDS/AML are recognized in the World Health Organization classification, based on the causative therapeutic exposure: alkylating agent/radiation or topoisomerase II inhibitor.14
Alkylating Agent/Radiation-Related Therapy-Related Myelodysplasia/Acute Myeloid Leukemia
In patients exposed to alkylating agents, t-MDS/AML usually appears 4 to 7 years after exposure. Approximately two-thirds of patients present with MDS and the remainder with AML with myelodysplastic features. Patients frequently present with cytopenias. There is a high prevalence of abnormalities involving chromosomes 5 (-5/del[5q]) and 7 (-7/del[7q]).
Topoisomerase II Inhibitor–Related Acute Myeloid Leukemia
AML secondary to topoisomerase II inhibitors presents as overt leukemia, without a preceding myelodysplastic phase. The latency is brief (6 months to 5 years) and is associated with balanced translocations involving chromosome bands 11q23 or 21q22. Other translocations include inv(18)(p13q22) or t(17,19)(q22;q12).15
Clinical Diagnosis
The common occurrence of cytopenias after autologous HCT and their nonspecific nature necessitated the need for specific criteria for diagnosing t-MDS/AML developing after HCT. These criteria include (1) significant marrow dysplasia in at least two cell lines, (2) peripheral cytopenias without alternative explanations, and (3) blasts in the marrow defined by French-American-British classification.16 In patients without blasts, presence of a clonal cytogenetic abnormality in addition to morphologic evidence of dysplasia may aid in making this diagnosis.
Risk Factors for Therapy-Related Myelodysplasia/Acute Myeloid Leukemia After Hematopoietic Cell Transplantation
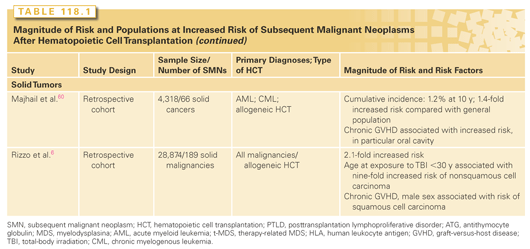
An increased risk of t-MDS/AML is associated with older age at HCT1; pretransplantation exposure to alkylating agents, topoisomerase II inhibitors, or radiation4; use of peripheral blood stem cells (PBSC; compared to bone marrow); stem cell mobilization with etoposide; conditioning with TBI; low number of CD34+ cells infused; and multiple transplants (see Table 118.1).1,4,17
The impact of pre-HCT chemotherapy and radiation on the risk of t-MDS/AML after HCT points toward an origin of events prior to HCT. A role for pretransplantation exposures is further supported by the observation that specific cytogenetic abnormalities observed posttransplantation have also been observed in the pretransplant marrow or peripheral blood graft among patients who develop t-MDS/AML after HCT, supporting a role for injury induced by prior cytotoxic chemotherapy in the etiology of t-MDS/AML.
TBI at 12 Gy does not increase the risk, whereas an increased risk is observed with TBI at 13.2 Gy.18 An association with TBI indicates that the disease may arise from residual stem cells that persist in the patient despite myeloablative treatment, rather than from re-infused stem cells, although TBI-induced alteration in the hematopoietic microenvironment may contribute to the development of t-MDS/AML.
Recipients of CD34-enriched cells isolated from peripheral blood after chemotherapy priming and growth factors are at a higher risk of t-MDS/AML as compared with recipients of CD34+ cells from the bone marrow.1,4 Potential explanations include harvesting of hematopoietic precursor cells damaged by chemotherapy at a time before they have completed DNA repair or an overrepresentation of damaged cells in the mobilized product. Supporting this hypothesis is the study4 demonstrating an increased risk of t-AML with 11q23 abnormalities among patients mobilized with high doses of etoposide for collection of stem cells prior to autologous HCT.
Patients receiving a smaller number of re-infused cells have been reported to be at an increased risk of t-MDS.17 Reconstitution of bone marrow clearly may result in greater proliferative stress in the setting of low stem cell numbers, increasing susceptibility to irreversible DNA damage associated with t-MDS. These observations are supported by in vitro data suggesting an increased proliferative stress placed on committed progenitors at the expense of primitive progenitors.19
Pathogenesis of Therapy-Related Myelodysplasia/Acute Myeloid Leukemia
t-MDS/AML is a clonal disorder that results from acquired genetic or epigenetic changes induced by cytotoxic therapy in hematopoietic stem cells. Improved understanding of the molecular pathogenesis of t-MDS/AML is required to allow development of strategies to identify populations at increased risk. Additionally, t-MDS/AML offers a unique perspective on mutagen-induced carcinogenesis. The clinical and epidemiologic data suggest that t-MDS/AML after autologous HCT results from genetic damage to the stem cell from pretransplant cytotoxic treatment, which may be potentiated by the transplant process itself.
Genetic Lesions Associated with Therapy-Related Myelodysplasia/Acute Myeloid Leukemia
Loss of chromosome 5 or del(5q) and loss of chromosome 7 or del(7q) are recurring abnormalities in t-MDS/AML. This has led to a search for candidate tumor suppressor genes in these regions. The majority of patients with 5q deletions exhibit losses at the 5q31 locus, with deletions in 5q33 being seen in some patients. Systematic evaluation of all 28 chromosome 5q31.2 genes for heterozygous nucleotide mutations or microdeletions using total exonic gene sequencing and array comparative genomic hybridization20 did not reveal any somatic nucleotide changes or cytogenetically silent 5q31.2 deletions. The mRNA levels of seven genes in the commonly deleted interval were reduced by 50% in CD34+ cells from del (5q) MDS samples, but complete loss of expression was not seen. These findings suggest that deletion leads to haploinsufficiency of multiple genes, but additional deletions or point mutations in 5q31.2 genes are uncommon. The role of specific genes has been evaluated. For example, the APC gene located in the commonly deleted 5q region has been studied. Mice with the Apcmin allele that results in a loss of APC function showed no abnormality in steady-state hematopoiesis but had reduced long-term stem cell function and developed an MDS phenotype over time,21 supporting a potential role for Apc haploinsufficiency in the MDS phenotype in patients with del (5q).
Chromosomal segment 7q22 is a common site of chromosome 7 deletions. Molecular analysis of a 2.5-Mb commonly deleted segment of chromosome band 7q22 has failed to uncover a candidate tumor suppressor gene. Mice with deletion of the targeted segment showed normal hematologic parameters and did not spontaneously develop myeloid malignancies.22 The targeted deletion did not cooperate with oncogenic Kras, Nf1 inactivation, or retroviral mutagenesis to accelerate leukemia development. These results fail to support the hypothesis that the 7q22 deletion contains a tumor suppressor gene. Further, 5q and 7q deletions in familial platelet disorder with leukemia are associated with mutations or deletions of a single AML1 allele.23 Balanced translocations involving the AML1 gene have also been associated with t-MDS/AML in patients exposed to topoisomerase II inhibitors.23 Transgenic expression of AML1-ETO leads to immortalization of murine myeloid progenitors but not overt leukemia; additional mutations are required for leukemogenesis.23 Therefore, altered AML1 function may have a major role in dysregulation of hematopoietic growth and genomic instability and predispose to leukemia through a multistep process.
Thus, although several candidate genes have been identified in these regions, including genes that regulate hematopoietic cell growth and differentiation, identification of a commonly deleted tumor suppressor gene has been elusive. It is possible that haploinsufficiency and reduced gene dosage for critical genes involved in hematopoiesis on 5q, including RPS14, EGR1, APC, NPM1, and CTNNA1, may sufficiently alter the balance between growth and differentiation to induce dysplastic hematopoiesis.24 Alternatively, epigenetic inactivation of the remaining allele or alterations in gene expression through loss of miRNA loci could play a role.
MLL rearrangements can be induced when primary human CD34+ cells are exposed to etoposide; stable genome rearrangements originating within the MLL translocation breakpoint hotspot were readily detected in etoposide-treated cells.25 The MLL gene located at chromosome band 11q23 has a major role in developmental regulation. Altered MLL function, as with AML1, may dysregulate hematopoietic growth and genomic instability and predispose to leukemia through a multistep process.
A second class of mutations in genes regulating cytokine signaling pathways has been recognized in AML. The RAS signaling pathway is activated downstream of several cytokine receptors. Constitutively activating point mutations of NRAS are detected in 10% to 15% of patients with t-MDS/AML. In contrast, mutations of the FLT3 gene (point mutations and internal tandem duplications), commonly seen in de novo AML, are not seen in t-MDS/AML.26
Genetic Susceptibility
The observed interindividual variability in risk of t-MDS/AML for a given genotoxic exposure is likely related to common polymorphisms in low-penetrance genes that regulate the availability of active drug metabolite, or those responsible for DNA repair. Knight et al.27 conducted a genome-wide association study in and identified three single nucleotide polymorphisms (rs1394384 [odds ratio (OR) = 0.3; 95% confidence interval (CI) = 0.2 to 0.6], rs1381392 [OR = 2.1; 95% CI = 1.3 to 3.4], and rs1199098 [OR = 0.5; 95% CI = 0.3 to 0.8]) to be associated with t-MDS/AML with chromosome 5/7 abnormalities.27 rs1394384 is intronic to ACCN1, a gene encoding an amiloride-sensitive cation channel that is a member of the degenerin/epithelial sodium channel; rs1199098 is in LD with IPMK, which encodes a multikinase that positively regulates the prosurvival AKT kinase and may modulate Wnt/beta-catenin signaling; rs1381392 is not near any known genes or regulatory elements. The investigators confirmed findings in an independent replication cohort.
Mouse strains resistant or susceptible to t-AML induced by the alkylator ethyl-N-nitrosourea were analyzed to identify genes that regulate t-AML susceptibility. Thirteen quantitative trait loci were significantly associated with ethyl-N-nitrosourea–induced leukemogenesis. These quantitative trait loci contained several genes that could contribute to leukemogenesis, including p53, DNA repair, and apoptosis-regulating genes, suggesting that susceptibility to alkylator-induced leukemia may be a complex trait related to multiple genes.28 The next section summarizes reports of associations between t-MDS/AML and low-penetrance genes that regulate the availability of active drug metabolite, or are responsible for DNA repair.
Drug Metabolism. Allan et al.29 examined associations between polymorphisms in the glutathione S-transferase genes (GSTM1, GSTT1, and GSTP1) and t-MDS/AML. Individuals with at least one GSTP1 codon 105 Val allele were significantly overrepresented in t-AML cases compared with de novo AML cases (OR = 1.8; 95% CI = 1.1 to 2.9). Also, relative to de novo AML, the GSTP1 codon 105 allele occurred more often among patients with t-MDS/AML with prior exposure to chemotherapy (OR = 2.7; 95% CI = 1.4 to 5.1), particularly among those with prior exposure to known GSTP1 substrates (OR = 4.3; 95% CI = 1.4 to 13.2) and not among patients with t-MDS/AML with exposure to radiation alone.
DNA Repair. Individuals with altered DNA repair mechanisms are likely susceptible to the development of genetic instability that drives the process of carcinogenesis as it relates to t-MDS/AML. Ellis et al.30 examined the association between patients with t-MDS/AML and two common functional p53-pathway variants: the MDM2 SNP309 and the TP53 codon 72 polymorphism. Neither polymorphism demonstrated a significant association. However, an interactive effect was detected such that MDM2 TT TP53 Arg/Arg double homozygotes, and individuals carrying both a MDM2 G allele and a TP53 Pro allele were at increased risk of chemotherapy-related t-MDS/AML.
Mismatch Repair. Defects in the mismatch repair pathway result in genetic instability or a mutator phenotype, manifested by an elevated rate of spontaneous mutations characterized as multiple replication errors in simple repetitive DNA sequences (microsatellites)—functionally identified as microsatellite instability (MSI). Approximately 50% of patients with t-MDS/AML have MSI, associated with methylation of the mismatch repair family member MLH1.31,32
Double-Strand Breaks. Cellular pathways available to repair double-strand breaks include homologous recombination, nonhomologous end-joining, and single-strand annealing. RAD51 is one of the central proteins in the homologous recombination pathway, functioning to bind to DNA and promote ATP-dependent homologous pairing and strand transfer reactions. RAD51-G-135C polymorphism is significantly overrepresented in patients with t-MDS/AML compared with controls (C allele: OR = 2.7).33 XRCC3 also functions in the homologous recombination double-strand break repair pathway by directly interacting with, and stabilizing RAD51. The variant XRCC3-241Met allele has been associated with a higher level of DNA adducts compared with cells with the wild-type allele, implying aberrant repair34 and has also been associated with increased levels of chromosome deletions in lymphocytes after exposure to radiation.35 Although XRCC3-Thr241Met was not associated with t-MDS/AML (OR = 1.4; 95% CI = 0.7 to 2.9), a synergistic effect resulting in an eight-fold increased risk of t-MDS/AML (OR = 8.1; 95% CI = 2.2 to 29.7) was observed in the presence of XRCC3-241Met and RAD51-135C allele in patients with t-MDS/AML compared with controls.33
Base Excision Repair. The XRCC1 protein plays a central role in the base excision repair pathway and also in the repair of single-strand breaks, by acting as a scaffold and recruiting other DNA repair proteins. The protein also has a BRCA1 C-terminus (BRCT) domain—a characteristic of proteins involved in DNA damage recognition and response. The presence of variant XRCC1-399Gln has been shown to be protective for t-MDS/AML.36
Nucleotide Excision Repair. Nucleotide excision repair removes structurally unrelated bulky damage induced by radiation and chemotherapy. The nucleotide excision repair pathway is linked to transcription and components of the pathway comprise the basal transcription factor IIH complex, which is required for transcription initiation by RNA polymerase II. One of the genes involved in the nucleotide excision repair pathway (ERCC2) is a member of the basal transcription factor IIH complex. The polymorphic Gln variant (ERCC2 Lys751Gln) is associated with t-MDS/AML.37
Telomeric Shortening
Telomeres are noncoding regions of DNA that provide a cap at the ends of chromosomes and prevent dicentric fusion and other chromosomal aberrations. Each somatic cell division is associated with a loss of telomere length. Cumulative telomere shortening can impose a limit on cell divisions and lead to cell senescence. Telomere shortening is also associated with genetic instability. Following HCT, the increased replicative demand on stem cells associated with hematopoietic regeneration can lead to accelerated telomere shortening. Telomere shortening could be an important issue in autologous HCT, especially when telomere length in the transplanted cells is already short because of prior chemotherapy, older age at HCT, or increased replicative stress on the stem cells because of a small number of cells transplanted. A prospective, longitudinal study revealed accelerated telomere shortening in patients with t-MDS/AML (preceding the onset of t-MDS/AML) when compared with matched controls who did not develop t-MDS/AML. Patients with t-MDS/AML also showed reduced generation of committed progenitors, suggesting that telomere alterations were related to reduced regenerative capacity of hematopoietic stem cells.38
Hematopoietic Abnormalities
Autologous HCT for NHL and HL is associated with hematopoietic abnormalities including marked and prolonged reduction in primitive progenitor long-term culture-initiating cells and committed progenitor colony-forming cell numbers, altered progenitor expansion potential, and microenvironmental defects. These abnormalities may be related in part to damage to hematopoietic cells from pretransplant chemotherapy because hematopoietic defects can also be seen in pretransplant samples.19 Although committed progenitors recover to pretransplant levels, primitive progenitor capacity is further depleted and does not show evidence of recovery for up to 2 years after HCT, consistent with extensive proliferation and differentiation of the committed progenitors and subsequent depletion of primitive progenitors during hematopoietic regeneration post-HCT.19 HCT may also be associated with defects in the marrow hematopoietic microenvironment, including reduction in stromal precursor growth and reduced capacity to support growth of myeloid progenitors and B progenitors. These microenvironmental defects may contribute to hematopoietic abnormalities posttransplantation. Extensive proliferation of stem cells bearing genotoxic damage posttransplant may have a role in establishment and amplification of an abnormal clone. Alternatively, the numerous replication cycles imposed on hematopoietic stem cells after HCT may result in excessive shortening of telomeres in descendent cells (as previously discussed). Telomeric shortening may be associated with genomic instability and chromosomal abnormalities, and could contribute to the pathogenesis of t-MDS/AML.39
CD34+ cells in the graft from patients with NHL also show reduced migration compared with normal CD34+ cells. In vitro migratory capacity of CD34+ cells in the graft correlated with the speed of hematopoietic recovery after transplantation.40 Therefore, pretransplant chemotherapy may also lead to reduced engraftment potential of primitive progenitor cells. HCT may also be associated with defects in the marrow hematopoietic microenvironment, including reduction in stromal precursor growth and reduced capacity to support growth of myeloid progenitors and B cell progenitors.41,42
Microenvironmental defects may contribute to hematopoietic abnormalities after transplantation. There is evidence supporting the importance of the hematopoietic microenvironment in MDS pathogenesis. Repeated sublethal irradiation of mice was shown to result in bone marrow dysfunction with features of MDS including low white blood cell counts, macrocytic anemia, and reduced platelets and megakryocytes. Bone marrow dysfunction was associated with increased bone marrow angiogenesis, vascular endothelial growth factor, and activation of the transcription factor NFkB. Tumor necrosis factor-α deleted mice were protected from irradiation effects,43 suggesting that radiation-mediated tumor necrosis factor-α induction may contribute to bone marrow dysfunction and t-MDS/AML. Deletion of the microRNA processing factor Dicer1 in mouse osteoblast progenitors resulted in hematopoietic abnormalities leading to development of MDS and AML, associated with reduced expression of the Sbds gene that is mutated in the Schwachman-Bodian-Diamond syndrome, an inherited human marrow failure disorder.44 Deletion of Sbds in osteoblast progenitors also induced marrow dysfunction and myelodysplasia. These results support a role for perturbation of microenvironmental cells in hematopoietic defects in t-MDS/AML.
Gene Expression Profiling
Gene expression profiling of acute lymphoblastic leukemia cells at diagnosis has been shown to be predictive of subsequent t-MDS/AML.45 Gene expression profiling of CD34+ hematopoietic progenitor cells from patients with t-MDS/AML has identified different subtypes of t-MDS/AML with characteristic gene expression patterns.46
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


