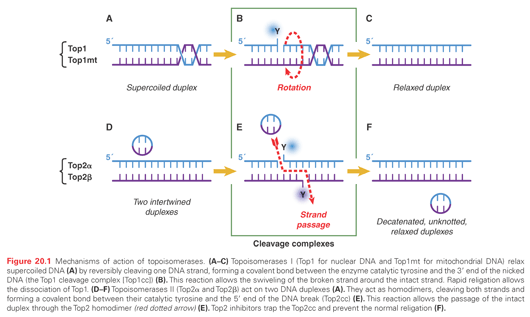
Biochemical Characteristics and Cleavage Complexes of the Different Topoisomerases
The DNA cutting/relegation mechanism is common to all topoisomerases and utilizes an enzyme catalytic tyrosine residue acting as a nucleophile and becoming covalently attached to the end of the broken DNA. These catalytic intermediates are referred to as cleavage complexes (see Fig. 20.1B, E). The reverse religation reaction is carried out by the attack of the ribose hydroxyl ends toward the tyrosyl-DNA bond.
Top1 (and Top1mt) attaches to the 3′-end of the break, whereas the other topoisomerases (Top2 and Top3) have opposite polarity and covalently attach to the 5′-end of the breaks (see Table 20.1 [second column] and Fig. 20.1B, E). Topoisomerases have distinct biochemical requirements. Top1 and Top1mt are the simplest, nicking/closing, and relaxing DNA as monomers in the absence of cofactor, and even at ice temperature. Top2 enzymes, on the other hand, are the most complex topoisomerases working as dimers, requiring ATP binding and hydrolysis, and a divalent metal (Mg2+) for catalysis. Top3 enzymes also require Mg2+ for catalysis but function as monomers without ATP requirement. Notably, the DNA substrates differ for Top3 enzymes. Whereas both Top1 and Top2 process double-stranded DNA, the Top3 substrates need to be single-stranded nucleic acids (DNA for Top3α and DNA or RNA for Top3β).10,12,13
Differential Topoisomerization Mechanisms: Swiveling Versus Strand Passage, DNA Versus RNA Topoisomerases
Topoisomerases use two main mechanisms to change nucleic topology. The first is by “untwisting” the DNA duplex. This mechanism is unique to Top1, which, by an enzyme-associated single-strand break, allows the broken strand to rotate around the intact strand (see Fig. 20.1B) until DNA supercoiling is dissipated. At this point, the stacking energy of adjacent DNA bases realigns the broken ends, and the 5′-hydroxyl end attacks the 3′-phosphotyrosyl end, thereby relegating the DNA. A remarkable feature of this Top1 untwisting mechanism is its extreme efficiency with a rotation speed around 6,000 rpm and relative independence from torque, thereby allowing full relaxation of DNA supercoiling.14
The second topologic mechanism is by “strand passage.” This mechanism allows the passage of a double- or a single-stranded DNA (or RNA) through the cleavage complexes. Top2α and Top2β both act by allowing the passage of an intact DNA duplex through the DNA double-strand break generated by the enzymes. After which, Top2 religates the broken duplex. Such reactions permit DNA decatenation, unknotting, and relaxation of supercoils.3 Top3 enzymes also act by strand passage but only pass one nucleic acid strand through the single-strand break generated by the enzymes. In the case of Top3α, the substrate is a single-stranded DNA segment (such as a double-Holliday junction), whereas in the case of Top3β, the substrate can be a single-stranded RNA segment, with Top3β acting as a RNA topoisomerase.13,15
TOPOISOMERASE INHIBITORS AS INTERFACIAL POISONS
Topoisomerase Inhibitors Act as Interfacial Inhibitors by Binding at the Topoisomerase–DNA Interface and Trapping Topoisomerase Cleavage Complexes
Relegation of the cleavage complexes is dependent on the structure of the ends of the broken DNA (i.e., the realignment of the broken ends). Binding the drugs at the enzyme–DNA interface misaligns the ends of the DNA and precludes relegation, resulting in the stabilization of the topoisomerase cleavage complexes (Top1cc and Top2cc). Crystal structures of drug-bound cleavage complexes have firmly established this mechanism for both Top1- and Top2-targeted drugs.16
It is critical to understand that the cytotoxic mechanism of topoisomerase inhibitors requires the drugs to trap the topoisomerase cleavage complexes rather than block catalytic activity. This sets apart topoisomerase inhibitors from classical enzyme inhibitors such as antifolates. Indeed, knocking out Top1 renders yeast cells totally immune to camptothecin,17,18 and reducing enzyme levels in cancer cells confers drug resistance. Conversely, in breast cancers, amplification of TOP2A, which is on the same locus as HER2, contributes to the efficacy of doxorubicin.19 Also, cellular mutations of Top1 and Top2 that renders cells insensitive to the trapping of topoisomerase cleavage complexes produce high resistance to Top1 or Top2 inhibitors. Based on this trapping of cleavage complexes mechanism, we refer to topoisomerase inhibitors as topoisomerase cleavage complex-targeted drugs.
Top1cc-Targeted Drugs (Camptothecin and Noncamptothecin Derivatives) Kill Cancer Cells by Replication Collisions
Top1cc are cytotoxic by their conversion into DNA damage by replication and transcription fork collisions. This explains why cytotoxicity is directly related to drug exposure and why arresting DNA replication protects cells from camptothecin.20,21 The collisions arise from the fact that the drugs, by slowing down the nicking/closing activity of Top1, uncouple the kinetics of Top1 with the polymerases and helicases, which lead polymerases to collide into Top1cc (Fig. 20.2A). Such collisions have two consequences. They generate double-strand breaks (replication and transcription runoff) and irreversible Top1–DNA adducts (see Fig. 20.2B). The replication double-strand breaks are repaired by homologous recombination, which explains the hypersensitivity of BRCA-deficient cancer cells to Top1cc-targeted drugs.22 The Top1-covalent complexes can be removed by two pathways, the excision pathway centered around tyrosyl-DNA-phosphodiesterase 1 (TDP1)23 and the endonuclease pathway involving 3′-flap endonucleases such as XPF-ERCC1.24 It is also possible that drug-trapped Top1cc directly generate DNA double-strand breaks when they are within 10 base pairs on opposite strands of the DNA duplex or when they occur next to a preexisting single-strand break on the opposite strand. Finally, it is not excluded that topologic defects contribute to the cytotoxicity of Top1cc-targeted drugs (the accumulation of supercoils25 and the formation of alternative structures such as R-loops) (see Fig. 20.2D).26

Cytotoxic Mechanisms of Top2cc-Targeted Drugs (Intercalators and Demethyl Epipodophyllotoxins)
Contrary to camptothecins, Top2 inhibitors kill cancer cells without requiring DNA replication fork collisions. Indeed, even after a 30-minute exposure, doxorubicin and other Top2cc-targeted drugs can kill over 99% of the cells, which is in vast excess of the fraction of S-phase cells in tissue culture (generally less than 50%).27,28 The collision mechanism in the case of Top2cc-targeted drugs (see Fig. 20.2A) appears to involve transcription and proteolysis of both Top2 and RNA polymerase II.29 Such situation would then lead to DNA double-strand breaks by disruption of the Top2 dimer interface (see Fig. 20.2C). Alternatively, the Top2 homodimer interface could be disjoined by mechanical tension (see Fig. 20.2C). Yet, it is important to bear in mind that 90% of Top2cc trapped by etoposide are not concerted and, therefore, consist in single-strand breaks,3,30,31 which is different from doxorubicin, which traps both Top2 monomers and produces a majority of DNA double-strand breaks.32 Finally, it is not excluded that topologic defects resulting from Top2 sequestration by the drug-induced cleavage complexes could contribute to the cytotoxicity of Top2cc-targeted drugs (see Fig. 20.2D). Such topologic defects would include persistent DNA knots and catenanes, potentially leading to chromosome breaks during mitosis.
TOPOISOMERASE I INHIBITORS: CAMPTOTHECINS AND BEYOND
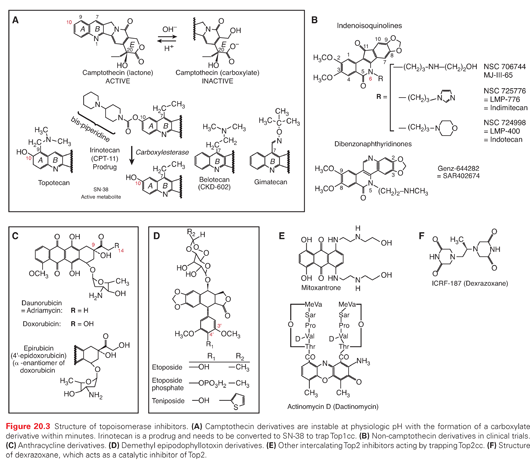
Camptothecin is an alkaloid identified in the 1960s by Wall and Wani33 in a screen of plant extracts for antineoplastic drugs. The two water-soluble derivatives of camptothecin containing the active lactone form are topotecan and irinotecan, which are approved by the U.S. Food and Drug Administration (FDA) for the treatment of several cancers. In addition, several Top1cc-targeting drugs are in clinical development, including camptothecin derivatives and formulations (including high–molecular-weight conjugates or liposomal formulations), as well as noncamptothecin compounds that exhibit greater potency or noncross resistance to irinotecan and topotecan in preclinical cancer models.31,34–36
Irinotecan
Irinotecan, a prodrug containing a bulky dipiperidine side chain at C-10 (Fig. 20.3), is cleaved by a carboxylesterase-converting enzyme in the liver and other tissues to generate the active metabolite, SN-38. Irinotecan is FDA approved for the treatment of colorectal cancer in the metastatic setting as first-line treatment in combination with 5-fluorouracil/leucovorin (5-FU/LV) and as a single agent in the second-line treatment of progressive colorectal cancer after 5-FU–based therapy (see Table 20.1).37,38 Newer therapeutic uses of irinotecan include a combination with oxaliplatin and 5-FU as first-line treatment in pancreatic cancer.39 Irinotecan is additionally used in combination with cisplatin or carboplatin in extensive-stage small-cell lung cancer40,41 as well as refractory esophageal and gastroesophageal junction (GEJ) cancers, gastric cancer, cervical cancer, anaplastic gliomas and glioblastomas, and non–small-cell lung cancer (Table 20.2). Irinotecan is usually administered intravenously at a dose of 125 mg/m2 for 4 weeks with a 2-week rest period in combination with bolus 5-FU/LV, 180 mg/m2 every 2 weeks in combination with an infusion of 5-FU/LV, or 350 mg/m2 every 3 weeks as a single agent.
Diarrhea and myelosuppression are the most common toxicities associated with irinotecan administration. Two mechanisms explain irinotecan-induced diarrhea. Acute cholinergic effects resulting in abdominal cramping and diarrhea occur within 24 hours of drug administration are the result of acetylcholinesterase inhibition by the prodrug, and can be treated with the administration of atropine. Direct mucosal cytotoxicity with diarrhea is typically observed after 24 hours and can result in significant morbidity. Symptoms are managed with loperamide. Hepatic metabolism and biliary excretion accounts for >70% of the elimination of the administered dose, with renal excretion accounting for the remainder of the dose. SN-38 is glucuronidated in the liver by UGT1A1, and deficiencies in this pathway increase the risk of diarrhea and myelosuppression. Dose reductions are recommended for patients who are homozygous for the UGT1A1*28 allele, for which an FDA-approved test for detection of the UGT1A1*28 allele in patients is available.42,43 Additionally, dose reductions of irinotecan are recommended for patients with hepatic dysfunction, with bilirubin greater than 1.5 mg/mL.44
Topotecan
Topotecan contains a basic side chain at position C-9 that enhances its water solubility (see Fig. 20.3). Topotecan is approved for the treatment of ovarian cancer,45 small-cell lung cancer,46 and as a single agent and in combination with cisplatin for cervical cancer.47 Additionally, it is active in acute myeloid leukemia (AML) and myelodysplastic syndrome (see Table 20.2). Topotecan is administered intravenously as a single agent at a dose of 1.5 mg/m2 as a 30-minute infusion daily for 5 days, followed by a 2-week period of rest for the treatment of solid tumors or at a dose of 0.75 mg/m2 as a 30-minute infusion daily for 3 days in combination with cisplatin on day 1, every 3 weeks, for the treatment of cervical cancer.
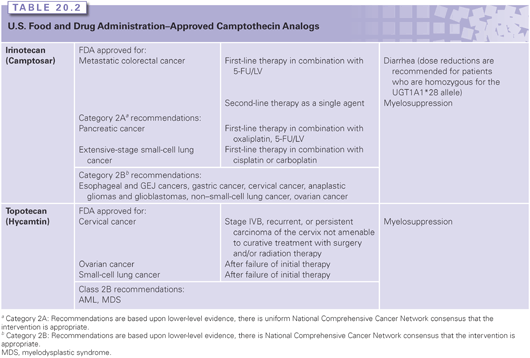
Myelosuppression is the most common dose-limiting toxicity. Extensive prior radiation or previous bone marrow–suppressive chemotherapy increases the risk of topotecan-induced myelosuppression. Other toxicities include nausea, vomiting, diarrhea, fatigue, alopecia, and transient hepatic transaminitis.
Topotecan and its metabolites are primarily cleared by the kidneys, requiring dose reduction in patients with renal dysfunction. A 50% dose reduction is recommended for patients with moderate renal impairment (creatinine clearance 20 to 39 mL per minute). There are no formal guidelines for dose reductions in patients with hepatic dysfunction (defined as serum bilirubin >1.5 mg/dL to <10 mg/dL). Topotecan additionally penetrates the blood-brain barrier, achieving concentrations in cerebrospinal fluid that are approximately 30% that of plasma levels.48
Camptothecin Conjugates and Analogs
New formulations of camptothecin conjugates and analogs are currently in clinical development in an effort to improve the therapeutic index (Table 20.3). The development of camptothecin conjugates is based on the notion that the addition of a bulky conjugate would allow for a more consistent delivery system and extend the half-life of the molecule.

CRLX101, formerly IT-101, a covalent cyclodextrin-polyethylene glycol copolymer camptothecin conjugate, has plasma concentrations and area under the curve (AUC) that are approximately 100-fold higher than camptothecin, with a half-life in the range of 17 to 20 hours compared to 1.3 hours for camptothecin.49 It has demonstrated antitumor activity in preclinical studies in irinotecan-resistant tumors with complete tumor regression in human non–small-cell lung cancer, Ewing sarcoma, and lymphoma xenograft models.50 Preliminary data from Phase 1 studies indicate that CRLX101 is well tolerated at a dose of 15 mg/m2 administered in a biweekly administration schedule.51 It is currently being studied in Phase 2 studies as a single agent and in combination with chemotherapeutic agents in lung, renal cell cancer, and gynecologic malignancies.52–54
Etirinotecan pegol (NKTR-102), an irinotecan polymer conjugate, has a longer plasma circulation time with a lower maximum concentration of SN-38 compared with irinotecan. It was evaluated in a Phase 2 study in platinum-resistant refractory epithelial ovarian cancer at a dose of 145 mg/m2 administered on a schedule of every 21 days; a median progression-free survival of 5.3 months and median overall survival of 11.7 months was observed.55 Two schedules of administration, 145 mg/m2 administered every 14 days versus every 21 days, have been tested in a Phase 2 study of NKTR-102 in patients with previously treated metastatic breast cancer.56 Of the 70 patients evaluated in this study, 20 patients achieved an objective response (29%; 95% confidence interval [CI] 18.4 to 40.6). For both these studies, the most common adverse events on the 21-day administration schedule were dehydration and diarrhea. Etirinotecan pegol is currently being evaluated in several phase 2 studies in lung cancer, colorectal cancer, and high-grade gliomas,57–60 with evidence of clinical activity in refractory solid tumors. A Phase 3 trial (The BEACON Study) is underway evaluating NKTR-102 against the physicians’ choice in refractory breast cancer.61
As an alternative to macromolecular conjugates, attempts have also been made to alter the camptothecin pentacyclic ring structure with modifications of the A and B ring (see Fig. 20.3A) in an effort to improve solubility and enhance antitumor activity. Structure–activity relationship studies have shown that substitutions at the 7, 9, and 10 positions serve to enhance the antitumor activity of camptothecin.62 Belotecan, a novel camptothecin analog, has a water-solubilizing group at the 7 position of the B ring of camptothecin (see Fig. 20.3A). Several Phase 2 studies have evaluated belotecan in combination with carboplatin in recurrent ovarian cancer63 and in combination with cisplatin in extensive-stage small-cell lung cancer,64 demonstrating activity in these cancers; however, these combinations were associated with prominent hematologic toxicities. Phase 2 studies evaluating belotecan as a single agent in patients with recurrent or progressive carcinoma of the uterine cervix failed to show activity.65 Gimatecan is a lipophilic oral camptothecin analog (see Fig. 20.3A). Pharmacokinetic studies demonstrate that gimatecan is primarily present in plasma as the lactone form (>85%), and has a long half-life of 77.1 +/− 29.6 hours, with an increase in maximum concentration (Cmax) and AUC of three- to six-fold after multiple dosing.66 Phase 2 studies show that gimatecan has demonstrated activity in previously treated ovarian cancer, with myelosuppression as the main toxicity.67
Newer development of analogs have attempted to modify the E-ring through introduction of an electron-withdrawing group at the α position in an effort to overcome the instability of the E-ring while maintaining the binding capability of the camptothecin analog to the Top1-DNA cleavage complex. Collectively called homocamptothecin analogs, two have been tested in clinical trials and include diflomotecan68 and elomotecan.69 The dose-limiting toxicity in the Phase I study of elomotecan was neutropenia. A five-member E-ring derivative has also been developed and has reached a Phase 1 clinical trial.70,71
Noncamptothecin Topoisomerase I Inhibitors
Noncamptothecin Top1 inhibitors are in clinical development, and include indenoisoquinolines and dibenzonaphthyridines (see Fig. 20.3B). Two indenoisoquinoline derivatives are currently in clinical development, indotecan (LMP400) and indimitecan (LMP776).72,73 Early in vitro studies show enhanced potency compared with camptothecins, and persistence of Top1 cleavage complexes.74 Genz-644282, a dibenzonaphthyridine derivative, demonstrated enhanced antitumor activity in preclinical studies75 and is currently being evaluated in Phase 1 clinical trials.76
TOPOISOMERASE II INHIBITORS: INTERCALATORS AND NONINTERCALATORS
Topoisomerase II inhibitors can be classified in two main classes: DNA intercalators, which encompass different chemical classes (Fig. 20.3C, E), and nonintercalators represented by the epipodophyllotoxin derivatives (see Fig. 20.3D). Although both act by trapping Top2 cleavage complexes (Top2cc), DNA intercalators exhibit a second effect as drug concentrations increase above low micromolar values: they block the formation of Top2cc by intercalating into DNA and destabilizing the binding of Top2 to DNA. This explains why Top2α and Top2β are trapped over a relatively narrow concentration range by anthracyclines, and why intercalators have additional effects besides trapping Top2cc, namely inhibition of a broad range of DNA processing enzymes including helicases, polymerase, and even nucleosome destabilization.
Doxorubicin
Doxorubicin and daunorubicin were the first anthracyclines discovered in the 1960s and remain among the most widely used anticancer agents over a broad spectrum of malignancies. Although doxorubicin only differs by one hydroxyl substitution on position 14 (see Fig. 20.3C), doxorubicin has a much broader anticancer activity than daunorubicin. Anthracyclines are natural products derived from Streptomyces peucetius variation caesius. They were found to target Top2 well after their clinical approval.77 Subsequent searches for less toxic drugs and formulations led to the approval of liposomal doxorubicin, idarubicin, and epirubicin.
Anthracyclines are flat, planar molecules that are relatively hydrophobic. The quinone structure of anthracyclines (see Fig. 20.3C) enhances the catalysis of oxidation-reduction reactions, thereby promoting the generation of oxygen free radicals, which may be involved in antitumor effects as well as the cardiotoxicity associated with these drugs.78,79 Anthracyclines are also substrates for P-glycoprotein and Mrp-1, and drug efflux is thought to be a major drug resistance determinant.80,81
Doxorubicin is available in a standard salt form and as a liposomal formulation. FDA-labeled indications for standard doxorubicin include acute lymphocytic leukemia (ALL), AML, chronic lymphoid leukemia, Hodgkin lymphoma, non-Hodgkin lymphoma, mantle cell lymphoma, multiple myeloma, mycosis fungoides, Kaposi sarcoma, breast cancer (adjuvant therapy and advanced), advanced prostate cancer, advanced gastric cancer, Ewing sarcoma, thyroid cancer, advanced nephroblastoma, advanced neuroblastoma, advanced non–small-cell lung cancer, advanced ovarian cancer, advanced transitional cell bladder cancer, cervical cancer, and Langerhans cell tumors. Doxorubicin has activity in other malignancies as well, including soft tissue sarcoma, osteosarcoma, carcinoid, and liver cancer (Table 20.4). Doxorubicin is typically administered at a recommended dose of 30 to 75 mg/m2 every 3 weeks intravenously.
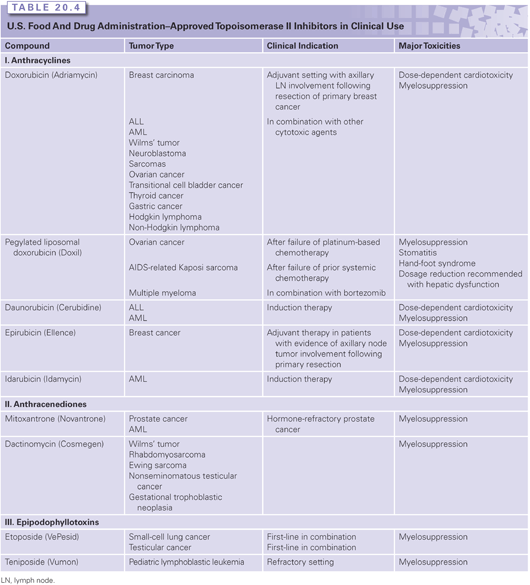
Major acute toxicities of doxorubicin include myelosuppression, mucositis, alopecia, nausea, and vomiting. Myelosuppression is the acute dose-limiting toxicity. Other toxicities, including diarrhea, nausea, vomiting, mucositis, and alopecia, are dose and schedule related. Prophylactic antiemetics are routinely given with bolus doses of doxorubicin, and longer infusions are associated with less nausea and less cardiotoxicity. Patients should also be warned to expect their urine to redden after drug administration. Doxorubicin is a potent vesicant, and extravasation can lead to severe necrosis of skin and local tissues, requiring surgical debridement and skin grafts. Infusions via a central venous catheter are recommended. Other toxicities of doxorubicin include radiation recall and the risk of developing secondary leukemia. Radiation recall is an inflammatory reaction at sites of previous radiation and can lead to pericarditis, pleural effusion, and skin rash. Secondary leukemias are thought to be a result of balanced translocations that result from Top2 poisoning by the anthracyclines, albeit to lesser degree than other Top2 poisons, such as the epipodophyllotoxins (see the following).82
Anthracyclines are cleared mainly by metabolism to less active forms and by biliary excretion. Less than 10% of the administered dose is cleared by the kidneys. Dose reductions should be made in patients with elevated plasma bilirubin. Doxorubicin should be dose reduced by 50% for plasma bilirubin concentrations ranging from 1.2 to 3.0 mg/dL, by 75% for values of 3.1 to 5.0 mg/dL, and withheld for values greater than 5 mg/dL.
Liposomal Doxorubicin
Doxorubicin is also available in a polyethylene glycol (PEG)ylated liposomal form, which allows for enhancement of drug delivery. Use of liposomal doxorubicin has been associated with less cardiotoxicity even at doses exceeding 500 mg/m2.83
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


