Tissue Factor and the Initiation and Regulation (TFPI) of Coagulation
James H. Morrissey
George J. Broze Jr.
Blood circulates as a liquid, but once it escapes from an injured blood vessel, it must rapidly gel, or clot, to form a hemostatic plug in order to limit further blood loss. On the other hand, pathologic activation of the blood clotting system within the lumen of blood vessels is a major cause of human death and disability. This chapter focuses on the mechanism of initiation of the blood clotting system, both in normal hemostasis and in many thrombotic diseases. Two main pathways for triggering the blood clotting cascade were initially described: the intrinsic pathway and the extrinsic, or tissue factor (TF), pathway. The intrinsic pathway is so called because it appeared to be an intrinsic property of plasma; that is, when blood or plasma is placed in a glass tube, it will clot spontaneously. This pathway is more accurately called the contact pathway because it is activated when plasma contacts artificial surfaces like glass or clay. The contact pathway per se is not thought to be of primary importance in normal hemostasis because individuals in whom it is inactive (owing to a severe congenital deficiency in factor XII) exhibit no bleeding tendency. The contact pathway is considered in detail in Chapter 15B.
The TF pathway of blood clotting is triggered when plasma comes into contact with cells that express the integral membrane protein, TF, on their surfaces. TF is abundant on a variety of cell types found outside the vasculature but is present only in tiny amounts within the vasculature. After vascular injury, blood is exposed to cells bearing TF, whereupon TF binds factor VII—one of the soluble clotting factors—with high affinity. It is the complex of TF and factor VII (or, more accurately, the activated form known as factor VIIa) that initiates the blood clotting cascade in normal hemostasis. In addition, TF triggers the clotting cascade in a variety of thrombotic disorders.
OVERVIEW OF THE INITIATION OF BLOOD COAGULATION BY TISSUE FACTOR AND FACTOR VII
The clotting cascade comprises a series of reactions involving the activation of zymogens (inert precursors of enzymes) by limited proteolysis. With the exception of thrombin, the newly generated proteases have low enzymatic activity until they bind to specific protein cofactors on suitable phospholipid (PL) surfaces. Such binding events potentiate the activity of clotting proteases by as much as several 100- to 1,000-fold.
The first enzyme in the clotting cascade consists of two subunits: a serine protease, factor VIIa (the catalytic subunit), and a protein cofactor, TF (the positively acting regulatory subunit). Because TF is an integral membrane protein, the complex of TF and factor VIIa (TF:VIIa) is tethered to the cell surface. Free factor VIIa is a weak enzyme, but TF:VIIa is the most potent known activator of the blood clotting cascade.
All the serine proteases of the blood clotting cascade, including factor VII, circulate in the plasma as inert zymogens. Furthermore, most protein cofactors of the clotting cascade circulate as inert procofactors. (TF is a notable exception; it does not require proteolysis for activity.) A nagging question for many years has been: How can the clotting cascade be initiated through the assembly of a collection of inert precursors? The answer appears to come from the fact that trace amounts of active factor VIIa are present in the circulation at all times.
The active forms of most serine proteases of the clotting system have extremely short plasma half-lives (measured in seconds) because plasma contains high concentrations of protease inhibitors. Free factor VIIa, however, is not reactive with plasma protease inhibitors.1 When injected into animals or humans, factor VIIa has a long circulating half-life of approximately 2 hours, similar to the approximately 5-hour half-life of zymogen factor VII.2 Therefore, any factor VIIa that is generated in vivo can circulate for considerable periods.
TF binds factor VII or VIIa with high affinity, resulting in a 1:1 complex of the two proteins on the cell surface. As depicted in FIGURE 11.1, once bound to TF, factor VII is rapidly converted to factor VIIa by limited proteolysis.3 So, there are two ways to form the TF:VIIa complex: through direct capture of circulating factor VIIa by TF or through capture of factor VII followed by conversion to factor VIIa.
Once formed, the TF:VIIa complex triggers clotting in two ways. TF:VIIa can activate factor IX via limited proteolysis (reaction 1 in FIGURE 11.2). The newly generated factor IXa then binds with a protein cofactor on a PL surface to form the IXa:VIIIa complex, which catalyzes the conversion of factor × to Xa. Alternatively, TF:VIIa can directly activate factor × (see reaction 2 in FIGURE 11.2). This latter mechanism predominates in most in vitro conditions, in which factor × is the preferred substrate for TF:VIIa.
Where does the first burst of serine protease activity come from if all the enzymes and their essential cofactors circulate as inactive precursors? It turns out that trace levels of factor VIIa are present in the circulation of all normal individuals, making up approximately 1% of the total factor VII concentration (but with considerable individual variation).4 Exposure of TF to plasma therefore results in assembly of both TF:VII and TF:VIIa complexes, and only the latter are enzymatically active. However, these low initial levels of TF:VIIa are thought to trigger enough of the clotting cascade to result in the back-activation of the rest of the bound factor VII by downstream proteases. A variety of coagulation proteases activate factor VII to VIIa in vitro, including factors IXa, Xa, and XIIa; thrombin; plasmin; and factor
VII-activating protease.3,5,6,7,8,9,10,11 In addition, the TF:VIIa complex catalyzes the autoactivation of factor VII bound to TF.12,13 It is currently unclear which of these proteases plays the major role in activating TF-bound factor VII during clotting, although many consider factor Xa the most likely candidate.
VII-activating protease.3,5,6,7,8,9,10,11 In addition, the TF:VIIa complex catalyzes the autoactivation of factor VII bound to TF.12,13 It is currently unclear which of these proteases plays the major role in activating TF-bound factor VII during clotting, although many consider factor Xa the most likely candidate.
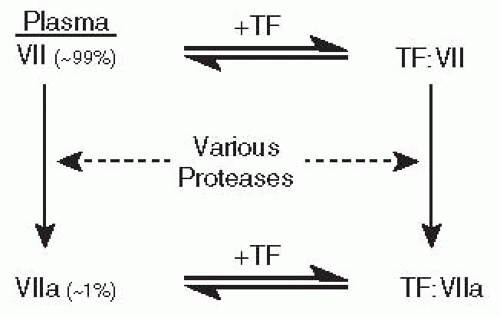 FIGURE 11.1 Two pathways for the formation of the TF and factor VIIa complex (TF:VIIa). Plasma contains a mixture of approximately 1% active factor VIIa and 99% zymogen factor VII. Both factors, VII and VIIa, bind reversibly to TF with similarly high affinities to generate TF:VII and TF:VIIa complexes, respectively. Factor VII can be converted to factor VIIa by a variety of plasma proteases (see text), and factor VII bound to TF is an especially good substrate for conversion to factor VIIa. |
It is also presently unclear where or how circulating factor VIIa is generated. One important clue comes from the discovery that patients with hemophilia B (who lack factor IX) have plasma factor VIIa levels that are only 10% of normal.14 Thus, factor IX (presumably as factor IXa) participates in formation of much of the circulating factor VIIa found in plasma. Another clue is that factor VIIa levels rise postprandially, especially after high-fat meals.15,16 This rise occurs in individuals who are deficient in factor XI or XII but not in factor IX,17 suggesting that activation of factor VII occurs on lipoprotein particles, apparently involving factor IXa. Consistent with this notion, plasma factor VIIa levels are very low in patients with familial abetalipoproteinemia.18 And finally, factor VIIa levels are higher in lymph than in plasma,19 suggesting that at least some of the factor VIIa may be generated extravascularly and returned to the circulation by the lymph.
TISSUE FACTOR PROTEIN STRUCTURE
TF is also known as thromboplastin, CD142, and coagulation factor III. It is a glycosylated membrane protein consisting of a single-polypeptide chain of 261 or 263 amino acids (the two forms are nearly equal in abundance), with variability in length owing to microheterogeneity at the amino-terminus.20,21,22 The calculated molecular weight of TF’s polypeptide chain is 29,447 Da or 29,593 Da, whereas the mobility of the fully glycosylated protein on sodium dodecyl sulfate (SDS) gels suggests a molecular weight of approximately 45,000 Da.23 TF belongs to the class 2 cytokine receptor superfamily.24 Topographically, TF is a type I integral membrane protein, with its amino-terminus located outside the cell and its carboxyterminus inside the cell. The overall domain structure of TF is given in FIGURE 11.3. The extracellular portion is composed of two fibronectin type III domains, which are a variation on the immunoglobulin fold.
The extracellular domain of TF has three N-linked carbohydrate chains,25 which were found to be completely dispensable for clotting activity since the recombinant protein produced in bacteria was fully functional.26 Recently, this conclusion was challenged by a study reporting that TF without carbohydrates had substantially reduced cofactor activity toward factor VIIa,27 although a following study by others reported no detectable role for the carbohydrate chains of TF in any of its functions, including coagulant activity, intracellular trafficking, or cellular signaling.28
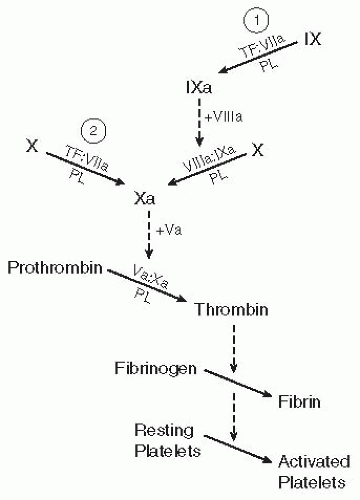 FIGURE 11.2 Simplified map of the blood clotting cascade initiated by the complex of TF and factor VIIa (TF:VIIa). The TF:VIIa complex can initiate clotting by activating either factor IX (reaction 1) or factor X (reaction 2). Regardless of the mechanism for triggering the clotting system, all clotting pathways converge at the formation of factor Xa, which assembles on a PL surface with its protein cofactor (factor Va) to catalyze the conversion of prothrombin to thrombin. Thrombin is responsible for converting fibrinogen to fibrin (again, by limited proteolysis), which spontaneously polymerizes to form a gel, or fibrin clot. In addition, thrombin is a potent activator of platelets (see Chapter 33). In this simplified clotting cascade, a number of back-activation reactions have been omitted. For example, factors Va and VIIIa circulate as inert precursors (factors V and VIII, respectively) and are activated following limited proteolysis by thrombin or factor Xa. The reactions involving hemostatically relevant factors XI and XIII, as well as all of the inhibitors of the clotting system, have also been omitted. |
The extracellular domain of TF has two disulfide bonds,29 at least one of which (the Cys186-Cys209 bond) is reported to be essential for coagulant activity,30 although this conclusion has been disputed.31 The Cys186-Cys209 disulfide bond has been proposed to play a role in TF encryption/decryption,32 although this conclusion has also been disputed31,33 (see section, Modulation of Factor VIIa Activity by Tissue Factor).
TF has a single membrane-spanning domain that anchors the protein to the cell surface. Membrane anchoring of TF is essential for full procoagulant activity, although the exact nature of the membrane anchor does not appear to be important for its clotting activity.34
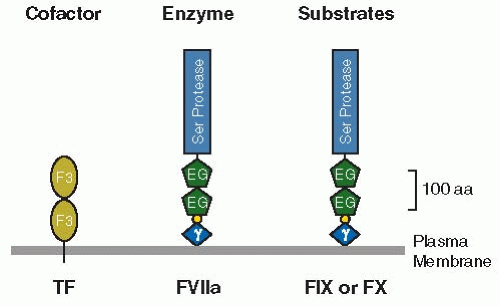 FIGURE 11.3 Schematic representation of the domain structure of TF, factor VII/VIIa (FVIIa), and factors IX and × (FIX and FX). The two fibronectin type III domains of TF are indicated as F3. The Gla domains of factors VII, IX, and × are indicated by γ, whereas the EGF-like domains are indicated by EG and the serine protease domains by Ser Protease. The aromatic stack is indicated by a small circle between the Gla and EGF domains. aa, amino acids. |
The short (23-amino acid long) cytoplasmic domain of TF contains a cysteine residue to which palmitate or stearate is attached by a thioester linkage.29 In addition, the cytoplasmic domain can be phosphorylated on serine.35 Deletion of the cytoplasmic domain has no discernible effect on TF procoagulant activity,34 making its role in blood clotting unclear, but there is increasing evidence that the cytoplasmic domain of TF may be important in some of TF’s nonhemostatic roles (see section, Nonhemostatic Functions for Tissue Factor and Factor VII).
TF has been purified from human brain and placenta using factor VII affinity chromatography23,36 and immunoaffinity chromatography.37,38 Larger quantities can be obtained as the recombinant protein expressed in eukaryotic or prokaryotic cells.26 Purified TF can readily be reconstituted into PL vesicles by using a variety of techniques.39,40,41 A soluble form of TF consisting of the isolated extracellular domain42 has proved useful in structure/function studies because it is easy to produce and handle.
Several groups have solved x-ray crystal structures of human and rabbit soluble TF.43,44,45,46,47 Soluble TF is an elongated and apparently rather rigid molecule in which the two fibronectin type III domains are joined at an angle of approximately 120 degrees. In addition, the x-ray crystal structures of the complex of soluble TF and factor VIIa have also been determined (discussed in the section, Structure of the Tissue Factor and Factor VIIa Complex). Further reference to crystal structures of TF and factor VII may be found at http://www.tf7.org/xray.htm.
SITES OF TISSUE FACTOR EXPRESSION: NORMAL AND PATHOLOGIC
TF, being an integral membrane protein, is mainly found on the surface of cells in which it is synthesized. TF is abundant in a variety of cell types distributed throughout the body, including adventitial cells surrounding all blood vessels larger than capillaries; differentiating keratinocytes in the skin; and a number of epithelial cell types, including those present in mucous membranes and many organ capsules.48,49,50 The distribution of TF can be rationalized by the requirement that it be present throughout the body, ready to trigger the clotting cascade at any time and place following vascular injury. This pattern of expression has been described as constituting a protective “hemostatic envelope” surrounding the vasculature, organ structures, and the entire organism.48 In addition, TF is abundant at anatomic sites in which the risk—or the deleterious consequences—of bleeding is high, such as the renal glomerulus and throughout the brain.48,50
Spontaneous bleeding in patients with hemophilia occurs most commonly in anatomic locations such as skeletal muscle and joints, which contain especially low levels of TF.48 Patients with hemophilia may bleed at these sites because direct activation of factor × by low levels of TF:VIIa is insufficient for hemostasis, and they lack the amplification step provided when TF:VIIa activates factor IX. Conversely, patients with hemophilia do not tend to bleed excessively from superficial cuts, which may be explained by the abundance of TF in the skin—exposure of large amounts of TF should allow direct activation of factor × in quantities sufficient for hemostasis.
In normal blood vessels, TF is readily detectable only in the adventitial cells within the vessel wall; TF antigen or messenger ribonucleic acid (mRNA) is usually undetectable in vascular smooth muscle and endothelial cells.48,49,50 Atherosclerotic plaques, however, contain significant levels of TF mRNA and antigen, associated with monocytes and smooth muscle cells within the plaque.49,51,52,53,54 TF antigen is also present in the acellular core of atheromas, most likely derived from cells that have undergone necrosis. TF in plaques can bind factor VIIa and is fully functional.53,54 In healthy arteries, TF is separated from the lumen by both the intima (endothelium and underlying basement membrane) and the media (multiple layers of vascular smooth muscle cells). In atherosclerosis, however, blood is separated from TF by only a thin monolayer of endothelial cells—a much more dangerous situation. It is generally accepted that myocardial infarction is triggered by rupture or fissure of atherosclerotic plaques in coronary arteries,55 exposing TF to blood within the lumen of the artery and triggering the formation of a thrombus that may occlude the vessel. Similar mechanisms likely precipitate other forms of arterial thrombosis, such as ischemic stroke.
TF is expressed in a variety of malignancies, where it can lead to thrombotic disease (e.g., in Trousseau syndrome). This can be caused by the expression of TF by the tumor cells themselves, by infiltrating monocytes, or by stromal cells. The association of TF with malignancy and its role in tumor pathology is reviewed in detail56,57 and in Chapter 125.
Experiments using specific inhibitors of TF:VIIa have demonstrated that coagulopathies associated with sepsis and septic shock are mediated by TF, and furthermore that TF:VIIa contributes directly to mortality in sepsis.58,59 Mice engineered to express very low levels of TF were protected against coagulopathy and mortality following challenge with bacterial endotoxin, and this was true even when low TF expression was limited to hematopoietic cells.60 These studies underscore the importance of monocyte TF in driving the life-threatening coagulopathy of sepsis.
TISSUE FACTOR GENE: STRUCTURE AND REGULATION
Located on chromosome 1 at p21-22,22,61 the TF gene (locus abbreviation, F3) spans 12.4 kb and contains six exons interrupted by five introns.62 The first exon contains the
5′ noncoding sequence of the mRNA and also encodes the signal peptide, which directs the protein to the membrane and which is removed during biosynthesis. The first exon is located within a CpG island, a GC-rich deoxyribonucleic acid (DNA) region approximately 1 kb long that contains an unusually high density of CpG codons. The next four exons encode the extracellular domain, whereas the sixth exon encodes the transmembrane and cytoplasmic domains. The sixth exon also contains the relatively long 3′ noncoding sequence, which is unusual because it includes an Alu-family repeat sequence that persists in the mature mRNA. The mRNA for TF is approximately 2.3 kb long, although in some cell types such as monocytes, minor amounts of a larger TF mRNA species (˜3.1 kb) are also observed.22 The longer mRNA retains the first intron, and it is thought not to encode an active protein.63,64
5′ noncoding sequence of the mRNA and also encodes the signal peptide, which directs the protein to the membrane and which is removed during biosynthesis. The first exon is located within a CpG island, a GC-rich deoxyribonucleic acid (DNA) region approximately 1 kb long that contains an unusually high density of CpG codons. The next four exons encode the extracellular domain, whereas the sixth exon encodes the transmembrane and cytoplasmic domains. The sixth exon also contains the relatively long 3′ noncoding sequence, which is unusual because it includes an Alu-family repeat sequence that persists in the mature mRNA. The mRNA for TF is approximately 2.3 kb long, although in some cell types such as monocytes, minor amounts of a larger TF mRNA species (˜3.1 kb) are also observed.22 The longer mRNA retains the first intron, and it is thought not to encode an active protein.63,64
Another alternatively spliced form of TF mRNA, initially discovered in certain leukemia cells, lacks the 5th exon.65 When translated, it would encode a TF protein lacking a portion of the extracellular domain (normally encoded by exon 5) that is replaced by a novel amino acid sequence derived from reading exon 6 out of frame. Bogdanov et al.66 have identified this alternatively spliced TF mRNA in a variety of normal cell types and have also discovered that the protein product of this alternatively spliced TF mRNA can be detected at low levels in plasma as an apparently soluble protein (discussed in the section, Blood-Borne Tissue Factor).
The TF gene is inducible in a variety of cultured cells. Quiescent fibroblasts in the dermis have no detectable TF antigen,48 and serum-starved fibroblasts in culture also do not express TF. When cultured quiescent fibroblasts are stimulated with serum or with various mitogenic cytokines, transcription of the TF gene is rapidly induced.67,68
Expression of TF on activated monocytes and macrophages is an important effector function of these cells in inflammation. Monocytes isolated from peripheral blood usually have undetectable levels of TF but express this protein strongly after stimulation with various inflammatory mediators.69,70 This directly parallels the observation that TF is expressed in circulating monocytes following intravenous administration of endotoxin or bacteria.71 Furthermore, leukocytes (in particular, monocytes) have been shown to mediate the Schwartzman reaction in endotoxin-stimulated animals.72 As with fibroblasts, the primary level of regulation of TF gene expression in monocytes is transcriptional.73 In addition, endotoxin treatment of monocytes prolongs the half-life of TF mRNA.63
In cell culture, vascular endothelial cells can be induced to express TF robustly in response to a variety of inducers, including interleukin-1, tumor necrosis factor-α, and bacterial endotoxin.69,70 In vivo, however, TF is expressed during severe sepsis by endothelial cells in only a few, highly restricted areas, such as in the splenic microvasculature.74 This suggests that additional factors, which are not necessarily replicated in vivo during sepsis, may govern TF expression in cultured endothelial cells. Endothelial expression of TF has been observed in vivo in other conditions, however, including placental villitis75 and graft rejection.76,77
In addition to studies with fibroblasts, monocytes, and endothelial cells, induction of TF gene expression has been studied in other cell types, including vascular smooth muscle cells and keratinocytes.69,70 A variety of agents that are capable of inducing TF expression have been identified.78 Transcriptional control of TF gene expression has been studied in several cell types, including monocytic cells, fibroblasts, and vascular endothelial and smooth muscle cells. These studies have identified transcription factor-binding sites flanking the promoter of the TF gene: Sp1 sites are important in basal transcription of the TF gene, whereas Egr-1 sites, AP-1 sites (binding c-Fos/c-Jun heterodimers), and a nuclear factor kappa B site (binding c-Rel/p65 heterodimers) are important in inducible expression of TF in response to bacterial endotoxin, serum, or phorbol ester, depending upon the cell type.79 TF gene expression is also induced by hypoxia in several cell types.80
FACTOR VII PROTEIN STRUCTURE
Factor VII, a glycosylated plasma protein, consists of a single polypeptide chain of 406 amino acids with an overall molecular weight of approximately 50,000 Da.7,81,82 Synthesized by the liver, it circulates in plasma at a concentration of about 500 ng/mL (10 nM).83 The domain structure of factor VII is depicted in FIGURE 11.3. Starting with the amino-terminus of the protein, it consists of the γ-carboxyglutamic acid-rich domain (Gla domain), the hydrophobic or aromatic stack, two epidermal growth factor (EGF)-like domains, and the serine protease domain. Molecular cloning revealed that factor VII has the same domain structure as factors IX, X, and protein C; shares considerable sequence homology with these coagulation serine proteases; and has a similar gene organization of introns and exons84,85 (see Chapter 9).
Typical of vitamin K-dependent coagulation proteins, factor VII is synthesized with a signal peptide on its amino-terminus that directs the protein to the endoplasmic reticulum for secretion, followed by a propeptide that directs the cell to modify glutamic acid residues in the Gla domain. The signal and propeptides are removed before secretion. Two types of complementary DNA clones for human factor VII have been identified, one of which has an additional 22 amino acids in the propeptide.84 Products of alternative splicing, these two forms of factor VII mRNA encode identical mature protein and appear to be functionally equivalent.
Factor VII contains 10 γ-carboxyglutamic acid (Gla) residues in its Gla domain, resulting from vitamin K-dependent modification of glutamic acid residues during biosynthesis. As with other vitamin K-dependent coagulation proteins, these Gla acid residues are essential for activity. Specifically, the Gla domain confers on factor VII the ability to bind, in a reversible and Ca2+-dependent manner, to membranes containing negatively charged PLs. In the crystal structure of the TF:VIIa complex, the Gla domain contains seven bound Ca2+,86 although three of these Ca2+-binding sites are probably actually occupied by Mg2+ in vivo.87
The aromatic stack of factor VII is a short amino acid sequence containing a small cluster of aromatic residues. It is adjacent to the Gla domain and, although encoded by a separate exon, is classified by some investigators as a part of the Gla domain. The two EGF domains have a typical growth factorfold and disulfide-bonding pattern. The first EGF domain binds a single Ca2+ with relatively high affinity,86,88 whereas the second EGF domain does not bind Ca2+. The first EGF domain of bovine factor VII contains the modified amino acid, β-hydroxyaspartic acid, whereas this modification is absent in human factor VII.89
The serine protease domain of factor VII is homologous to the digestive enzymes trypsin and chymotrypsin and exhibits
trypsin-like substrate specificity (i.e., cleaving peptide bonds after arginine or lysine). It has a typical serine protease catalytic triad composed of His 193, Asp242, and Ser344. As with many other coagulation serine proteases, the protease domain of factor VII binds a single Ca2+.86,90 The protease domain also binds zinc ions, which inhibit factor VIIa enzymatic activity.87,91,92
trypsin-like substrate specificity (i.e., cleaving peptide bonds after arginine or lysine). It has a typical serine protease catalytic triad composed of His 193, Asp242, and Ser344. As with many other coagulation serine proteases, the protease domain of factor VII binds a single Ca2+.86,90 The protease domain also binds zinc ions, which inhibit factor VIIa enzymatic activity.87,91,92
Zymogen factor VII is converted to factor VIIa, the enzymatically competent form, by proteolysis of a single peptide bond between Arg152 and Ile153. The result, factor VIIa, is composed of two disulfide-linked polypeptide chains. The light chain of factor VIIa, consisting of the Gla domain, aromatic stack, and both EGF domains, has 152 amino acids and a molecular weight of approximately 20,000 Da. The heavy chain, consisting of the serine protease domain, has 254 amino acids and a molecular weight of approximately 30,000. Unlike many other coagulation zymogens, factor VII does not release an activation peptide when converted to factor VIIa.
Factor VII contains two N-linked carbohydrate chains attached to residues Asn145 and Asn322.89 It also contains two short, O-linked carbohydrate chains attached to the first EGF domain: glucose, glucose-xylose, or glucose-(xylose)2 attached to Ser52; and a single fucose attached to Ser60.93
Factor VII can be purified from plasma using conventional chromatography7,81,82 or immunoaffinity chromatography.94 Recombinant human factor VII has been produced in cultured mammalian cells.89 Truncated forms of factor VII have been produced in which the Gla domain has been removed (Gladomainless factor VII) by limited proteolysis95,96 or by expression of the truncated recombinant protein.97 Gla-domainless factor VII may or may not contain the aromatic stack, depending on the site of cleavage. Furthermore, a form of factor VII lacking both the Gla domain and the first EGF domain has also been produced recombinantly,98,99,100 and it fails to bind to TF.98 Factor VII mutants have been produced, in which the activation cleavage site has been mutated so that it cannot be activated by the usual activators,101 which demonstrated that zymogen factor VII had no detectable enzymatic activity. In addition, the active site serine (Ser344) has been mutated to alanine, generating a form of factor VIIa without enzyme activity which has been useful in investigating the conversion of factor VII to VIIa.12
STRUCTURE OF THE TISSUE FACTOR AND FACTOR VIIA COMPLEX
The x-ray crystal structure of the complex of soluble TF and active site-inhibited factor VIIa has been solved to a resolution of 2.0 Å,86 and the structure of a similar complex inhibited with a modified version of bovine pancreatic trypsin inhibitor has been solved to a resolution of 2.1 Å.102 The structure of TF in these complexes is very similar to that of soluble TF alone, confirming the relative rigidity of this molecule. Within the complex, factor VIIa is a highly elongated protein that forms extended contacts with TF (see FIGURE 11.4). The main sites of contact on factor VIIa are located in the first EGF domain and the protease domain, with additional points of contact within the second EGF domain and the aromatic stack region of the Gla domain. The sites of contact on TF are located on both fibronectin type III domains and the interfacial region between them.
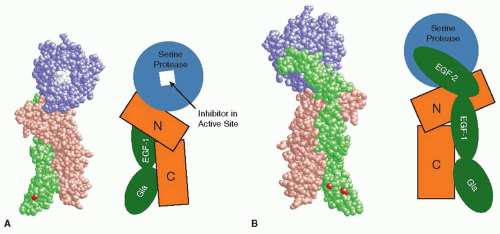 FIGURE 11.4 Two views of the x-ray crystal structure of the complex of soluble TF and active site-inhibited factor VIIa (left) and diagrammatic representations of the domain organizations of TF and factor VIIa (right). A: “Front” view of the TF:VIIa complex, with the active site facing the viewer. B: “Back” view of the complex, with the active site facing away from the viewer. The membrane surface would be located at the bottom of each complex. TF is shaded brown, whereas factor VIIa is shown in green (light chain) and blue (heavy chain). In addition, the tripeptidylchlormethylketone inhibitor in the active site of factor VIIa is colored white and calcium ions are red. For the TF representation, N and C refer to the N-terminal and C-terminal fibronectin type III domains, respectively. For the factor VIIa representation, the Gla domain is labeled Gla and the EGF-like domains are labeled EGF-1 or EGF-2. The serine protease domain and the inhibitor are also labeled. The two views of the TF:VIIa complex were generated with the program RASMOL,424 using the atomic coordinates file 1DAN86 from the Protein Data Bank. |
Several studies have used domain swapping and site-directed mutagenesis to identify the binding sites on TF and factor VIIa that permit them to interact with each other,103,104,105,106 revealing that some contacts are important for stabilizing the binding of TF to factor VIIa (chiefly with the first EGF domain of factor VIIa), whereas others appear to be important in enhancing the enzymatic activity of factor VIIa (chiefly with the protease domain of factor VIIa). These findings are reviewed in detail elsewhere.105,106
The portion of the Gla domain located at the bottom of the factor VIIa molecule (as shown in FIGURE 11.4) contains three solvent-exposed hydrophobic residues that are proposed to be inserted into the hydrophobic core of the PL bilayer.107 The carboxy-terminus of soluble TF is also located near the bottom of the structure, as presented in FIGURE 11.4; in intact TF, this part of the molecule is linked to the membrane anchor by a short peptide sequence. Therefore, in the views given in FIGURE 11.4, the membrane surface should be located at the bottom of the complex, with TF linked to the membrane by its membraneanchoring domain and with VIIa interacting with PL head groups through the Gla domain. Fluorescence resonance energy-transfer experiments indicate that the TF:VIIa complex is oriented almost perpendicular to the membrane, with the active site of factor VIIa located approximately 78 Å above the membrane surface.108,109 This same orientation was observed in detailed molecular dynamics simulations of the TF:VIIa complex on membranes.110
MODULATION OF FACTOR VIIA ACTIVITY BY TISSUE FACTOR
When factor VIIa binds to TF, its ability to catalyze the hydrolysis of small peptidyl-amide or peptidyl-ester substrates is enhanced 20-fold to 100-fold, depending upon the substrate.42,111,112,113 This is usually accomplished by an increase in the catalytic rate constant (kcat) and, in some cases, by a decrease in the Michaelis-Menten constant (Km), indicating that TF is an allosteric activator of factor VIIa.
Substrate hydrolysis by serine proteases is a multistep process,114 and the mechanism by which TF enhances the enzymatic activity of factor VIIa is not completely understood. Studies with a slowly hydrolyzed model substrate have shown that TF enhances the rate of substrate hydrolysis by factor VIIa chiefly by enhancing substrate affinity of factor VIIa rather than by altering the rates of any of the subsequent catalytic steps.115
A wealth of structure-function studies combining site-directed mutagenesis and x-ray crystallography have enhanced the understanding of the allosteric activation of factor VIIa by TF.116,117 Insights into the mechanism of allosteric activation of factor VIIa have also been obtained by designing mutants of factor VIIa that have enhanced enzymatic activity in the absence of TF.118,119 One of the most surprising results comes from the crystal structure of zymogen factor VII, which exhibits extensive β-strand re-registration relative to the crystal structures of factor VIIa.100
Serine protease zymogens are converted to active enzymes by limited proteolysis, whereupon the newly generated amino-terminus folds back into the protein to stabilize the oxyanion pocket.120 It has been proposed that free factor VIIa exists in two states in equilibrium with each other: one in which the protein is catalytically active and a second, zymogen-like state, in which the amino-terminus of the heavy chain is not properly folded back into the catalytic domain.121,122 Surprisingly, in the crystal structure of factor VIIa obtained in the absence of TF or any inhibitor in the active site, the amino-terminus of the factor VIIa heavy chain was still folded back into the catalytic domain and the oxyanion pocket was intact.99
Free factor VIIa proteolytically activates factors IX and × extremely slowly, but when it binds to TF that has been incorporated into suitable PL vesicles, its activity is enhanced many 1,000-fold.123,124,125 (Factors IX and × will bind to PL vesicles only if they contain negatively charged PLs, most particularly phosphatidylserine; therefore, TF is most often reconstituted into vesicles containing a mixture of phosphatidylserine and the neutral PL, phosphatidylcholine.) There are three main reasons for this rate enhancement. First, the reversible binding of factors IX and X to PL surfaces raises the concentration of these substrates in the vicinity of the enzyme TF:VIIa, which reduces the apparent Km. Second, TF is an allosteric activator of factor VIIa enzymatic activity. Third, TF appears to provide an extended binding site (exosite) for macromolecular substrates, as identified by site-directed mutagenesis.97,126,127
The fact that most coagulation reactions are restricted to two-dimensional surfaces substantially complicates the interpretation of their enzyme kinetics.128,129,130 In addition, coagulation reactions can occur in vivo under conditions of stasis or under conditions of considerable blood flow. Andree and Nemerson131 have published extensively on the effects of flow on the kinetics of factor × activation by the TF:VIIa complex.
It has long been known that resting, intact cells have much lower procoagulant activity than do cells that have been damaged, lysed, or treated with calcium ionophore.132 Although present on the surface of intact cells, TF becomes fully active only when the membrane properties of the cell are altered,133,134 a process that is sometimes referred to as decryption of “encrypted” cell-surface TF. There are several potential explanations for TF encryption/decryption. The first is that most cells restrict the distribution of aminophospholipids (such as phosphatidylserine) to the inner leaflet of the plasma membrane.135 Exposure of negatively charged PLs on the outer leaflet is necessary for efficient binding of substrate (i.e., factor IX or X), so the membrane asymmetry of intact cells will tend to limit the activity of TF. When cells are lysed, damaged, or treated with calcium ionophore, PL asymmetry is lost, resulting in the exposure of phosphatidylserine and phosphatidylethanolamine on the outer leaflet of the plasma membrane, thereby enhancing the activity of TF:VIIa. Another potential explanation in some cell types is that TF may associate with caveolae, areas of the cell surface with altered lipid composition.136,137 It has been also proposed that dimerization or oligomerization of TF in the membrane may reduce its activity and that damage or lysis of cells may foster the formation of TF monomers with enhanced activity.138
Finally, it has been proposed that Cys186 and Cys209 of TF participate in an unusual “allosteric” disulfide bond that can readily be broken or re-formed (i.e., reduced or reoxidized) to switch TF from a predominantly signaling form to a highly procoagulant form on cell surfaces.139 It has further been proposed that extracellular protein disulfide isomerase (PDI) may regulate TF encryption/decryption via this allosteric Cys186-Cys209 bond, and also derivatization of these Cys residues via S-nitrosylation or glutathionation.32 These proposals have been disputed by others, in reports that the Cys186-Cys209 bond of
TF is not required for coagulant activity and that PDI appears not to play a direct role in TF encryption/decryption.31,33,140
TF is not required for coagulant activity and that PDI appears not to play a direct role in TF encryption/decryption.31,33,140
TF:VIIa has an extremely restricted substrate specificity; its main natural substrates appear to be factors VII, IX, and X. Factor × is the favored substrate under most in vitro conditions, although activation reactions taking place on intact cells can show a preference for either factor IX or factor X125,141,142,143 for reasons that are not clear. When factor IX is activated, two peptide bonds must be cleaved. TF:VIIa complex catalyzes the cleavage of one of these bonds (i.e., at Arg180) more efficiently than the other (i.e., at Arg145), and, furthermore, factor Xa and TF:VIIa complex can synergize to activate factor IX to IXa more efficiently.144
Factor VII is a substrate for factor VIIa145 in an autoactivation reaction enhanced by TF.12 This reaction requires that the TF:VIIa and TF:VII complexes encounter each other by lateral diffusion in the plane of the membrane.13 Autoactivation of factor VII might be important in initiating the clotting cascade, but its precise role is yet to be elucidated.
TISSUE FACTOR PATHWAY INHIBITOR
In 1947, Thomas148 and Schneider149 showed that blood contained an inhibitor of TF-initiated coagulation. Ten years later, Hjort150 described many of the in vitro properties of the inhibitor, but it took an additional 30 years before the inhibitor was isolated.151 Previously called antithromboplastin, anticonvertin, TF inhibitor, extrinsic pathway inhibitor, or lipoprotein-associated coagulation inhibitor, in 1991 the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis settled on the currently used name, tissue factor pathway inhibitor (TFPI).
STRUCTURE OF TFPI ISOFORMS
The TFPI gene spans about 90 kb on the long arm of chromosome 2 (q32) and contains 10 exons (GenBank: NC_000002.11).152,153 All the splice junctions between exons are of the same type (type I), suggesting that the TFPI gene was assembled during evolution through a process of gene duplication and exon shuffling. Two alternatively transcribed isoforms of TFPI have been identified. TFPIα (GenBank NM_006287.4) is the originally isolated form of TFPI. The mature TFPIα protein contains an acidic amino-terminus, followed by three tandem Kunitz-type protease inhibitory domains, and a basic carboxyterminus (FIGURE 11.5). With posttranslational modifications, including N-and O-linked glycosylation and partial phosphorylation of Ser2, the molecular mass of TFPIα is approximately 43 kDa.154,155 In TFPIβ (GenBank NM_001032281.2), an alternative carboxyterminus is inserted at residue 182, which directs the attachment of a glycosyl phosphatidylinositol (GPI) anchor (FIGURE 11.6). The protein mass of TFPIβ is less than that of TFPIα (22 vs. 32 kDa), but it migrates on SDS-PAGE at the same apparent molecular weight as TFPIα due to additional O-linked carbohydrate.156 In human umbilical vein endothelial cells and endothelial-like cell lines (EAhy926, ECV304), the ratio of TFPIα/TFPIβ mRNA varies between 5 and 10.157
Kunitz-type inhibitors act by a standard mechanism in which the inhibitor feigns to be a good substrate, but, after the enzyme binds, subsequent cleavage between the P1 and P1′ amino acid residues of the inhibitor occurs slowly or not at all. The P1 residue is an important determinant of the specificity and activity of these inhibitors. In kinetic terms, Kunitz-type inhibitors produce slow, tight-binding, competitive, and reversible enzyme inhibition. In the reaction, the Kunitz inhibitor forms an initial “encounter” complex (EI) with the enzyme, and this complex then “slowly” isomerizes to a much tighter form (EI*). “Slow” implies that the final degree of inhibition does not occur immediately and “tight-binding” means that the inhibition occurs at a concentration of the inhibitor that is near to that of the enzyme being inhibited. The Kunitz-2 domain of TFPI mediates factor Xa binding and inhibition, whereas the Kunitz-1 domain is necessary for the inhibition of factor VIIa in the TF:VIIa complex.158 The Kunitz-3 domain of TFPIα lacks protease inhibitory activity.159 TFPI also inhibits trypsin and chymotrypsin reasonably well and inhibits cathepsin G, plasmin, and activated protein C (APC) poorly; the physiologic significance of these other inhibitory reactions, however, is obscure.
DISTRIBUTION OF TFPI
The microvascular endothelium is felt to be the major source of TFPI in vivo. Northern blot analysis shows the highest TFPI mRNA levels in the placenta and lung and the lowest in the brain.160 Studies of normal tissues have detected TFPI protein in the endothelium of the microvasculature, smooth muscle cells, monocytes/macrophages, megakaryocytes/platelets, mesangial cells, fibroblasts, microglia, cardiomyocytes, and mesothelial cells.
Plasma TFPI
The plasma concentration of TFPI is approximately 1.6 nM (˜70 ng/mL). Most of the circulating TFPI is bound to lipoproteins (˜80%), and manipulations that alter the levels of low-density lipoprotein (LDL), a major carrier of TFPI, also affect TFPI levels.161,162,163 TFPI circulating with LDL is 34 kDa and appears to represent a substantially truncated form, which lacks a large portion of Kunitz-3 domain and the C-terminal domain of TFPIα.161,164 The 41 kDa form of TFPI that is bound to high-density lipoprotein seems to represent a similar carboxy-truncated form that is linked in a mixed disulfide bond with monomeric apolipoprotein-AII. Whether the lipoprotein-associated TFPIs are extensively truncated forms of TFPIα or modified forms of TFPIβ has not been established, and the mechanism underlying the association of 34 kDa with LDL has not been determined. Nonlipoprotein-bound or “free” TFPI is 20% of total plasma TFPI and is a combination of forms of TFPIα with limited carboxyterminal truncation produced by an as yet to be unidentified protease(s) (10%) and full-length TFPIα (10%). The concentration of full-length TFPIα is reduced in the plasmas of patients with factor V or protein S deficiency, and binding interactions between full-length TFPIα and these proteins have been demonstrated.165,166 Whether these interactions affect the expression, proteolytic degradation, or clearance of TFPIα remains to be determined. Oral contraceptives and hormone replacement therapy reduce plasma levels of TFPI.167,168,169,170
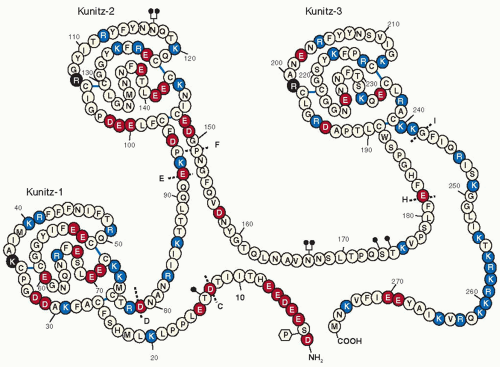 FIGURE 11.5 Structure of TFPIα. Amino acids are identified by the single-letter code (encircled). Positively charged amino acids are shown in red, negatively charge amino acids in blue, and neutral amino acids in white (histidine residues are considered uncharged). The Kunitz domains are labeled and the basic P1 residues at the active site cleft for each domain is shown in black. N-linked glycosylation sites are denoted at N117 and N167 by  and O-linked glycosylation sites are denoted at T14, S174, and T175 by and O-linked glycosylation sites are denoted at T14, S174, and T175 by  . Partial phosphorylation at Ser2 is also shown . Partial phosphorylation at Ser2 is also shown  . The sites of introns in the TFPI gene are labeled with dotted lines and capital letters. (Figure created by Thomas Girard.) . The sites of introns in the TFPI gene are labeled with dotted lines and capital letters. (Figure created by Thomas Girard.) |
Platelet TFPI
TFPIα is expressed by megakaryocytes, stored in platelets at a site separate from α granules, and released in response to thrombin and other agonists.171,172 In “coated” platelets produced by dual agonist stimulation (e.g., thrombin/collagen), a proportion of the released TFPIα remains bound and functional at the platelet surface. Similar to other ligands detected on the surface of coated platelets, transglutaminase inhibitors prevent this retention of surface TFPIα.172 The full-length TFPIα carried by platelets is 8% to 10% of the total TFPI in blood, which is comparable to the quantity of soluble full-length TFPIα in plasma. Therefore, it is likely that platelets are a major source of full-length TFPIα, the most anticoagulantly active form of TFPI (see below), at local sites of coagulation where platelets aggregate. On the other hand, polyphosphates released from the dense granules of platelets reduce the anticoagulant effect of TFPIα in plasma coagulation assays, apparently in part through their enhancement of factor V activation.173,174
Cell-Associated TFPI
Several reports have documented the low-affinity, heparin-inhibitable binding of TFPIα to cells, the internalization and degradation of TFPIα through the action of LDL-receptor-related protein (LRP), and the rapid clearance of TFPIα in animals.98,175,176,177,178,179,180,181,182,183 These studies used recombinant TFPIα (rTFPIα) that lacked the posttranslational modifications found in mammalian TFPIα. In contrast, the rTFPIα produced by a mammalian cell line (mouse C127) does not bind to cells or interact with LRP in the same fashion as Escherichia coli rTFPIα and is cleared from plasma at a 10-fold slower rate.184 Therefore, the studies that used nonmammalian-derived TFPI should probably be viewed with considerable caution.
A substantial fraction of the TFPI produced by endothelial cells remains at the cell surface, associates with caveolae, and is released by phosphatidylinositol-specific phospholipase C (PIPLC). These are properties of GPI-anchored proteins. The PIPLC-induced release of TFPIβ is consistent with its possession of an intrinsic GPI anchor.157 It has been reported that
cell-surface TFPIα is also released by PIPLC treatment.185,186 Since the amino acid sequence of TFPIα does not contain the canonical motifs to direct the attachment of a GPI anchor, this implied that the cell-surface binding of TFPIα may involve its interaction with a separate GPI-anchored coreceptor(s), which controlled its cellular trafficking and surface expression.187 Subsequent work, however, has shown that it is TFPIβ that is released by PIPLC from endothelial cells and placental microsomes.188 In endothelial cells induced to produce TF, GPI anchorage of TFPI serves to redistribute TF:VIIa to caveolae in a factor Xa-dependent manner.136,185,189 GPI anchorage, however, may not be a requirement for TF:VIIa inhibition by cell-surface TFPI as chimeric forms of rTFPI anchored by GPI or transmembrane domains produce similar levels of TF:VIIa inhibition.190 Endothelial cell-associated TFPI represents a substantial reservoir of TFPI in vivo, suggesting that the regulation of TF-mediated procoagulant and cell-signaling activities in the endothelium is a critical physiologic role of TFPI. Whether TFPI is attached to the surface of other TFPI-expressing cells (e.g., vascular smooth muscle cells, monocytes/macrophages) in the same manner as it is in endothelial cells remains to be determined.
cell-surface TFPIα is also released by PIPLC treatment.185,186 Since the amino acid sequence of TFPIα does not contain the canonical motifs to direct the attachment of a GPI anchor, this implied that the cell-surface binding of TFPIα may involve its interaction with a separate GPI-anchored coreceptor(s), which controlled its cellular trafficking and surface expression.187 Subsequent work, however, has shown that it is TFPIβ that is released by PIPLC from endothelial cells and placental microsomes.188 In endothelial cells induced to produce TF, GPI anchorage of TFPI serves to redistribute TF:VIIa to caveolae in a factor Xa-dependent manner.136,185,189 GPI anchorage, however, may not be a requirement for TF:VIIa inhibition by cell-surface TFPI as chimeric forms of rTFPI anchored by GPI or transmembrane domains produce similar levels of TF:VIIa inhibition.190 Endothelial cell-associated TFPI represents a substantial reservoir of TFPI in vivo, suggesting that the regulation of TF-mediated procoagulant and cell-signaling activities in the endothelium is a critical physiologic role of TFPI. Whether TFPI is attached to the surface of other TFPI-expressing cells (e.g., vascular smooth muscle cells, monocytes/macrophages) in the same manner as it is in endothelial cells remains to be determined.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


