46.1
Introduction
Thyroid hormone is essential for systemic regulation of metabolism, growth, and development, and homeostatic control of thyroid status is indispensable for the maintenance of health and fitness. Bone and cartilage are key targets for thyroid hormone action, and thyroid hormones have major effects on the developing skeleton and directly affect the regulation of bone mass and strength in adults. Bone strength and fracture susceptibility are determined by acquisition of peak bone mass in early adulthood and the rate of subsequent bone loss with aging. Euthyroid status is essential for optimal bone mineral accrual during the development and regulation of bone turnover in adults. Population epidemiological studies demonstrate that abnormal thyroid status is associated with an increased risk of osteoporotic fragility fracture, whether it is due to thyrotoxicosis, subclinical hyperthyroidism, thyroid status at the upper end of the normal reference range, or overtreatment of hypothyroidism .
Thyroid disease is common with a prevalence that rises with increasing age and is higher in women. The prevalence of hypothyroidism in females is between 1% and 2%, and the lifetime incidence of hyperthyroidism is 0.5%–2% . Subclinical hypo- or hyperthyroidism, in which thyroid-stimulating hormone (TSH, thyrotropin) is respectively raised or suppressed despite normal thyroid hormone levels, is even more common and may affect up to 10% of women over the age of 55. Hyperthyroidism is most commonly caused by the autoimmune condition Graves’ disease but also results from toxic multinodular goiter or isolated toxic nodules in the thyroid gland and can be transient resulting from subacute thyroiditis. Causes of thyroiditis include viral or bacterial infections, postpartum or silent immune-mediated inflammation, and drugs or radiation. Hypothyroidism also most commonly results from an autoimmune inflammatory condition, termed Hashimoto’s thyroiditis. Thyroid hormone replacement with levothyroxine is the mainstay of treatment for overt hypothyroidism, although some patients with subclinical hypothyroidism may also be treated with levothyroxine.
A number of rare heritable conditions caused by mutations in genes involved in thyroid hormone metabolism and signaling have important consequences in the skeleton. Many of these mutations have been replicated in mouse models, leading to major advances in our understanding of the mechanisms of thyroid hormone action in bone and cartilage .
46.2
Systemic regulation of thyroid hormone action
Circulating thyroid hormone levels are maintained in the normal range by classical negative feedback regulation. Thyrotropin-releasing hormone (TRH) is synthesized in the paraventricular nucleus of the hypothalamus and acts on the anterior pituitary gland to stimulate secretion of TSH. TSH acts on the G-protein coupled TSH receptor to stimulate the growth of thyroid follicular cells and synthesis and release of thyroid hormones. Thyroid hormone levels are maintained at a genetically determined set point of the hypothalamic–pituitary–thyroid (HPT) axis by acting on nuclear thyroid hormone receptors (TRs) in the hypothalamus and pituitary to inhibit secretion of TRH and TSH, respectively . Thus circulating TSH and thyroid hormone levels are maintained in a physiological inverse relationship ( Fig. 46.1 ) .
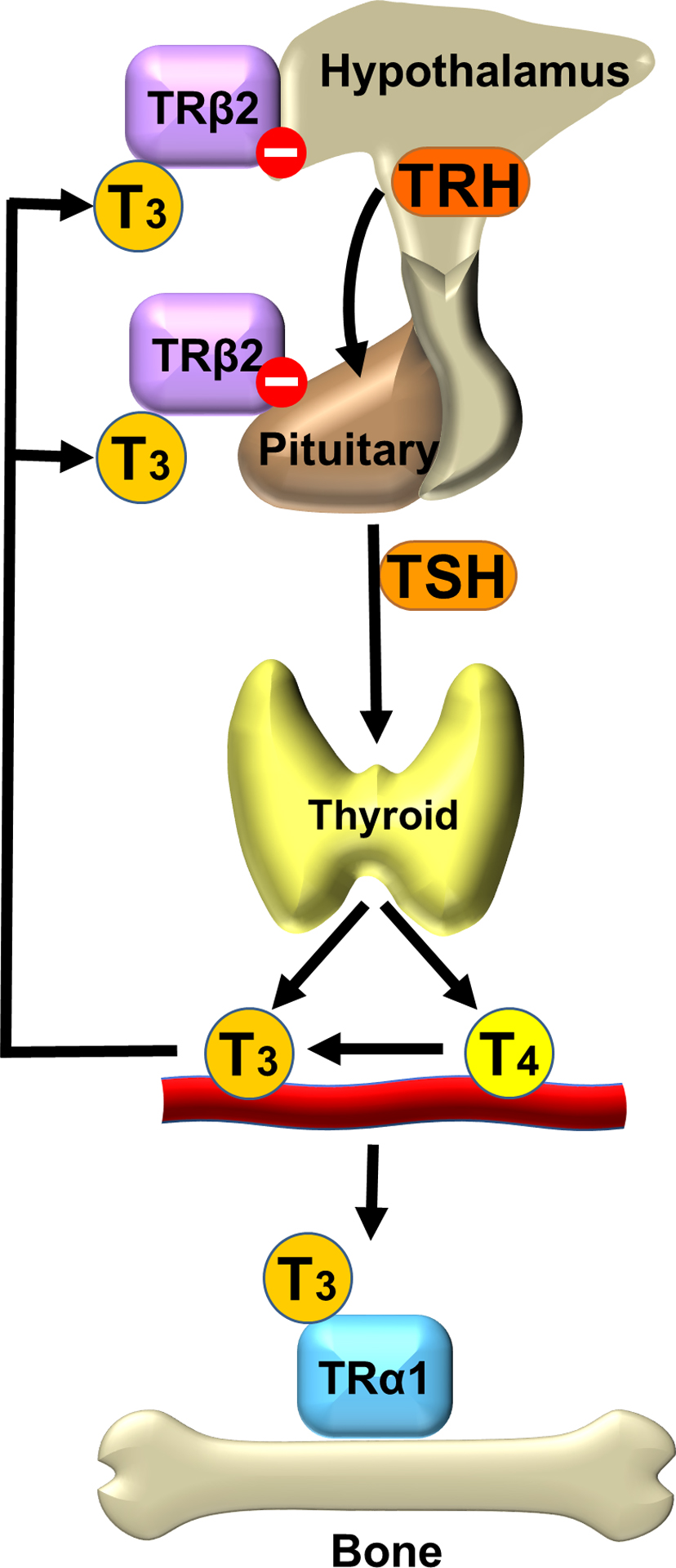
The thyroid gland mainly synthesizes the prohormone thyroxine (T4) together with a small amount of the active hormone 3,5,3′- l -triiodothyronine (T3) in a ratio of 10:1, while approximately 80% of the total daily T3 production is generated by peripheral conversion from T4 by the type 1 and 2 iodothyronine deiodinases (DIO1 and DIO2) . Consequently, the ratio of serum free T4 to free T3 is approximately 3:1 .
46.3
Cellular transport and metabolism of thyroid hormone
Both T4 and T3 are actively transported across the cell membrane by high-affinity-specific transporters, including monocarboxylate transporter-8 (MCT8), MCT10, organic acid transporter protein-1c1 (OATP1c1), and the organic anion transporter SLC17A4 . Availability of T3 to the nuclear TR in T3 target tissues is dependent on intracellular conversion of T4 to T3 by DIO2 and inactivation of T3 by the type 3 deiodinase (DIO3). DIO2 catalyzes removal of the 5′-iodine atom from the outer ring of the prohormone T4 to generate the active hormone T3. DIO3, however, catalyzes removal of the inner ring 5-iodine atom, converting T3 to the inactive metabolite 3,3′-diiodothyronine (T2) and preventing activation of T4 by converting it to 3,3′,5′-triiodothyronine (rT3) . Overall, the ratio of the activities of the DIO2 and DIO3 enzymes within T3 target tissues determines the local intracellular concentration of T3 that is available to activate the TR in the nucleus.
46.4
Thyroid hormone receptors
T3 binds with high affinity to TRs that form heterodimers with retinoid X receptors and bind to thyroid hormone response elements in the promoter regions of T3-regulated target genes ( Fig. 46.2 ) . TRs are encoded by two genes THRA and THRB that encode the functional receptors TRα1, TRβ1, and TRβ2, respectively . TRα1 and TRβ1 are widely expressed, though their patterns of expression vary between tissues and at different stages of embryonic development . TRβ2 is the main TR isoform that mediates negative feedback inhibition of TRH and TSH in the hypothalamus and pituitary .
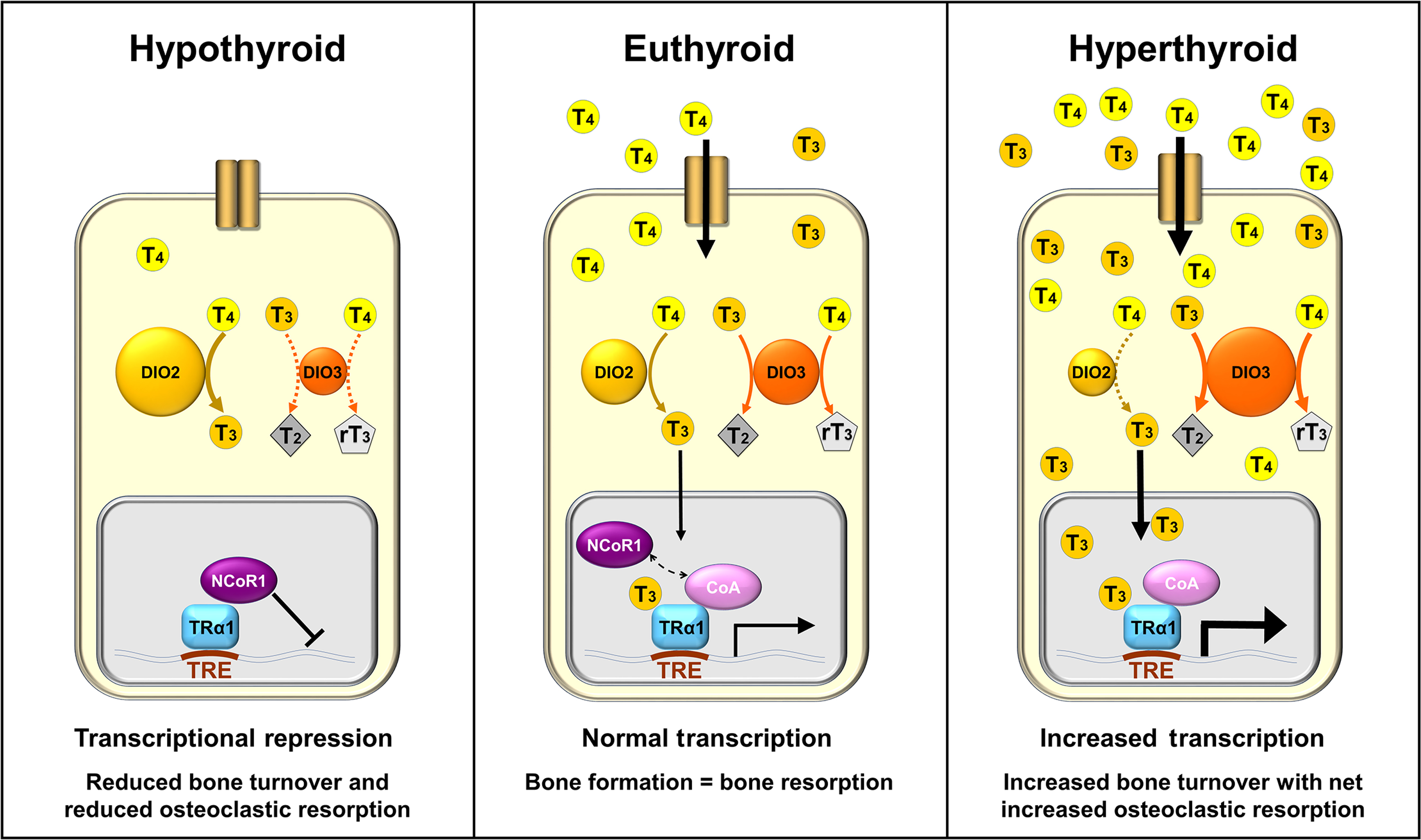
TR complexes regulate target gene transcription by recruiting coactivator or corepressor proteins, which are interchanged in response to binding of T3 ligand . These coregulating proteins induce histone modifications and changes in chromatin structure that activate or repress transcription of target genes . In the absence of T3, TRs act predominantly as transcriptional repressors of target gene expression, mediated via corepressors. In the hyperthyroid state, high levels of T3 cause pathologically increased target gene expression, mediated via coactivators. In the euthyroid state, gene expression is activated in a concentration-dependent manner in response to T3 .
46.5
Thyroid hormone action in bone
The effects of T3 on the skeleton are predominantly mediated by the nuclear actions of TRα1, which stimulate T3 target gene expression directly , but both TR isoforms are expressed in growth plate chondrocytes, bone marrow stromal cells, and osteoblasts. TRα1 is expressed at much higher levels than TRβ1 in bone , though studies in mice also suggest physiological roles for TRβ1 during endochondral ossification and in the adult skeleton . Thyroid hormone transporters and the deiodinases DIO2 and DIO3 are expressed in chondrocytes and osteoblasts, where they regulate uptake and intracellular availability of T3 .
T3 promotes hypertrophic differentiation of chondrocytes and mineralization of cartilage necessary for skeletal development. Long bones, vertebrae and ribs, are formed by endochondral ossification in which chondrocytes differentiate from mesenchymal progenitor cells and form a cartilage anlage that provides a mineralized scaffold for bone formation . Long bone formation initiates in primary ossification centers located in the diaphyses via coordinated processes of chondrocyte proliferation, differentiation, cartilage mineralization, and apoptosis. Longitudinal bone growth subsequently occurs via a similarly organized process of endochondral ossification in the epiphyseal growth plates that are located at the ends of the long bones between the primary ossification center and the secondary ossification centers that form the epiphyses .
T3 regulates chondrocyte differentiation in the growth plates, acting together with growth hormone, insulin-like growth factor-1, leptin, fibroblast growth factors (FGFs), and parathyroid hormone-related peptide (PTHrP) via downstream signaling pathways, including Wnt/beta-catenin, to stimulate expression of bone morphogenic protein 4, Wnt4, and FGFR3 . Ultimately, the local actions of T3 in the growth plate determine the set point of a complex negative feedback loop, involving Indian hedgehog (IHH), PTHrP, and bone morphogenic protein receptor-1A, that regulates the pace of chondrocyte differentiation and the rate of linear growth .
At the base of the growth plate, hypertrophic chondrocytes undergo apoptosis or may transdifferentiate to the osteoblast lineage . T3 stimulates expression of aggrecanase-2, ADAMTS5, and matrix metalloproteinase-13 to facilitate cartilage matrix degradation and also stimulates IHH and osterix signaling resulting in transformation of hypertrophic chondrocytes toward bone matrix–producing osteoblasts . T3 further regulates bone matrix synthesis and mineralization by stimulating osteoblast proliferation and differentiation, acting via the critical Wnt-signaling pathway that is essential for bone formation .
Longitudinal growth proceeds until the growth plates fuse in puberty. Bone mass accrual continues into early adulthood, and peak bone mass is typically achieved after 20–30 years of age. The process of adult bone maintenance occurs via the continuous turnover of mineralized bone matrix . Bone turnover occurs within multicellular units of the bone remodeling compartment that comprise osteocytes, osteoclasts, and osteoblasts whose functions are tightly coordinated by paracrine signaling. Bone turnover is also dependent on regulation by systemic hormones, including thyroid hormones, which predominantly stimulate osteoclastic bone resorption. Thus in animal studies, increased thyroid hormone concentrations cause bone loss in both trabecular and cortical compartments due to a generalized but uncoupled increase in bone turnover that favors resorption over bone formation . Although thyroid hormone excess stimulates bone resorption, it is not certain whether T3 acts directly in osteoclasts. Mct8 , Dio2 and Dio3 mRNAs, and DIO3 enzyme activity have been detected in osteoclasts , and expression of TRα1 and TRβ1 mRNA has also been detected . Nevertheless, it remains unclear whether T3 indirectly stimulates bone resorption via primary TRα1-mediated actions in osteoblasts or stromal cells, or whether T3 acts directly in osteoclasts .
46.6
Action of thyroid-stimulating hormone receptor in bone
The TSH receptor is expressed primarily in thyroid follicular cells where it mediates HPT axis regulation of thyroid hormone secretion. Nevertheless, Tshr expression has also been detected in osteoblasts, osteoclasts, and chondrocytes , as well as in other tissues . TSHα and TSHβ subunits are not expressed in human or mouse osteoblasts or osteoclasts , indicating that TSH does not have autocrine effects in these cells. Thyrostimulin, an alternative ancestral thyroid stimulating hormone receptor or thyrotrophin receptor (TSHR) ligand, is, however, expressed in mouse osteoblasts and osteoclasts and studies of Gpb5 −/− mice that lack thyrostimulin suggested it may have a role in bone formation during development .
The role of TSH in skeletal cells has proved difficult to clarify as contradictory findings have been reported. Thus growth plate cartilage and cultured chondrocytes express TSHR, and treatment with TSH decreases expression of SOX9 and type IIa collagen in primary chondrocytes, suggesting that TSH inhibits chondrocyte differentiation . However, in another study, TSH increased markers of chondrocyte differentiation in mesenchymal stem cells .
Likewise, in initial studies, treatment of primary osteoblasts with TSH inhibited osteoblastogenesis and reduced expression of osteoblast cell markers , whereas, in subsequent studies, TSH stimulated osteoblast differentiation and function . In other studies, it was found that TSHR protein was expressed at low levels in osteoblasts. While TSH binding to the TSHR and consequent cAMP activation was reported in human osteoblasts, other studies found that TSH had no effect on osteoblast differentiation or function .
Osteoclasts have also been shown to express TSHR protein at low levels, and treatment with TSH and TSHR-stimulating antibodies also had no effect on osteoclast differentiation or function in some studies . Nevertheless, most studies have shown that TSH does inhibit osteoclast formation and function and, although underlying mechanisms and signaling pathways remain incompletely defined, inhibition of TNFα signaling has been proposed as an important downstream mediator of the TSH response.
Overall, these studies demonstrate that the TSHR is expressed at low levels in skeletal cells and is likely to be responsive to thyrostimulin and TSH. Nevertheless, the downstream effects of TSHR signaling in chondrocytes and osteoblasts remain unclear, whereas most studies in osteoclasts suggest that TSH has an inhibitory role.
46.7
Studies in genetically modified mice
46.7.1
Targeting thyroid hormone transporters
Genetically modified mice are a powerful tool to investigate the individual components of thyroid hormone transport, metabolism and action, and their contributions to skeletal physiology ( Table 46.1 ). Thyroid hormone transporters are necessary for the active transport of thyroid hormones into cells. Mice lacking the thyroid hormone transporter MCT8 have elevated T3 but decreased T4 levels . Mct8 y/− mice have only a minor delay in postnatal growth suggesting that other transporters may compensate for deficiency of MCT8 . Nevertheless, analysis of Mct8 knockout mice revealed delayed endochondral ossification due to impaired T3 signaling in chondrocytes, and Mct8 y/− mice also exhibited decreased bone mineralization and strength in adulthood . Although MCT8 is important for endochondral ossification and adult bone maintenance, other studies provide further evidence to suggest functional redundancy between thyroid hormone transporters in the skeleton. Thus although Mct10 mutant mice and Oatp1c1 −/− knockout mice have normal weight gain and linear growth, Mct8 and Oatp1c1 double knockout mice display significant postnatal growth retardation .
| Mouse model | Genotype | Thyroid status | Skeletal phenotype | Interpretation | References |
|---|---|---|---|---|---|
| Transporter mutants | |||||
| Mct8 −/− | No Mct8 | TSH 1–3× | Minor early growth delay; delayed endochondral ossification | Reduced T3 uptakes into chondrocytes and osteoblasts | |
| T4 0.3–0.7× | |||||
| T3 1–1.4× | |||||
| Mct10 Y88 * /Y88 * | No Mct10 | At P21: T4 normal, T3 0.8 | Normal growth | ||
| In adulthood: T4 and T3 normal | |||||
| Oatp1c1 −/− | No Oatp1c1 | TSH, T4 and T3 all normal | Normal growth | ||
| Mct8 −/− Oatp1c1 −/− | No Mct8 or Oatp1c1 | TSH 5× | Growth retardation after P16 | ||
| T4 0.3× | |||||
| T3 2× | |||||
| Deiodinase mutants | |||||
| Dio1 −/− | No DIO1 | TSH normal | Normal growth | DIO2 compensates | |
| T4 1.4× | |||||
| T3 normal rT3 4× | |||||
| Dio2 −/− | No DIO2 | TSH 3–15× | Normal intramembranous and endochondral ossification; in adulthood, increased mineralization, reduced bone formation and brittle bones | DIO1 compensates, but reduced T3 action in osteoblasts | |
| T4 1–1.3× | |||||
| T3 normal | |||||
| rT3 normal | |||||
| Dio3 −/− | No DIO3 | Perinatal thyrotoxicosis; adult central hypothyroidism | Reduced body length; adult skeletal phenotype not described | Reduced clearance of T3 | |
| TR mutants | |||||
| TRα 0/0 | No TRα | TSH, T4 and T3 all normal (euthyroid) | Transient growth delay, delayed endochondral ossification, reduced mineralization; in adulthood, osteosclerosis with increased trabecular bone volume | Reduced T3 action in bone | |
| Normal TRβ | |||||
| TRα1 PV/+ | Heterozygous dominant-negative TRα | TSH 1.5–2× | Severe persistent growth retardation, delayed endochondral and intramembranous ossification, impaired chondrocyte differentiation and reduced mineralization in development; in adulthood, grossly dysmorphic bones and osteosclerosis with increased trabecular and cortical bone volume, reduced osteoclastic bone resorption resistant to T4 treatment | Reduced T3 action in bone and dominant-negative basal action of unliganded TRα1 | |
| T4 normal | |||||
| T3 1.2× (Euthyroid) | |||||
| TRβ −/− | No TRβ | TSH ↑ | Advanced endochondral and intramembranous ossification in development and short stature; in adulthood, increased osteoclastic bone resorption and osteoporosis | Increased T3 action via TRα1 | |
| Normal TRα | |||||
| T4 ↑, T3 ↑ | |||||
| RTH and goiter | |||||
| TRβ PV/PV | Homozygous dominant-negative TRβ | TSH ↑↑ | Accelerated prenatal growth, postnatal growth retardation, advanced endochondral and intramembranous ossification, and increased mineralization in development | Increased T3 action via TRα1 | |
| T4 ↑, T3 ↑ | |||||
| Severe RTH and goiter | |||||
| TR corepressor mutants | |||||
| TRα1 PV/+ | Mutant nuclear corepressor of unliganded TR | TSH normal | Improvement of the abnormal skeletal phenotype of TRα1 PV/+ mice | NCoR mediates the basal actions of the unliganded TR in bone | |
| NCoR ΔID/ΔID | |||||
| T4 ↓ | |||||
| T3 normal (Abnormal HPT axis of TRα1 PV/+ mice reversed) | |||||
| Congenital hypothyroid | |||||
| Pax8 −/− | Maximal TSHR signaling | TSH ↑↑ | In development, delayed endochondral ossification, impaired linear growth, reduced bone mineralization; in adulthood, reduced cortical bone, reduced trabecular bone with impaired remodeling, reduced mineralization (majority die by weaning) | Effect of severe hypothyroidism on development | |
| T4 and T3 undetectable (no thyroid) | |||||
| Unliganded TRα and TRβ | |||||
| TSHR mutants | |||||
| Tshr −/− | No TSHR | TSH ↑↑ | Similar to Pax8 −/− mice above | Severe hypothyroidism; phenotype similar to Pax8 −/− mice despite absent TSH signaling | |
| Unliganded TRα and TRβ | T4 and T3 undetectable (hypoplastic thyroid) | ||||
| Tshr P556L/P556L (Tshr hyt/hyt ) | Absent TSHR signaling | TSH ↑↑ | Similar to Pax8 −/− mice above; in adulthood phenotype not described | Severe hypothyroidism; phenotype similar to Pax8 −/− mice despite absent TSH signaling | |
| Unliganded TRα and TRβ | T4 and T3 ↓↓ (hypoplastic thyroid) | ||||
46.7.2
Targeting thyroid hormone metabolism
The skeletal consequences of mutation or deletion of the deiodinase enzymes have also been investigated. Dio1 is not expressed in the skeleton and, accordingly, DIO1-deficient mice display normal weight gain and linear growth, although the skeleton has not been studied in detail .
Dio2 is expressed mainly in osteoblasts in the skeleton , and Dio2 −/− mice have a threefold increase in TSH, a small increase in T4 and normal T3 levels due to impaired deiodination of T4 in the pituitary . DIO2 deficiency and absent T4 to T3 conversion in osteoblasts in Dio2 −/− mice resulted in decreased bone turnover, increased bone mineralization and brittle bones in adults, whereas skeletal development and postnatal growth were normal . Nevertheless, mutant mice harboring a Thr92Ala polymorphism in DIO2, which has been associated with increased bone turnover markers and decreased BMD in humans, were recently shown to have normal linear growth and normal bone mineral content and strength . Taken together, these data demonstrate that DIO2 in osteoblasts is indispensable and essential for the maintenance of normal adult bone mass and strength, whereas a Thr92Ala polymorphism in DIO2 that results in minor biochemical changes in enzyme activity has no effect on bone mineralization and strength.
Finally, the role of DIO3 in bone has not been investigated specifically. Although Dio3 −/− mice have severe growth retardation, they also have a multisystem disorder and high perinatal mortality that confounds interpretation of the skeletal consequences following Dio3 deletion . Thus cell-specific gene targeting will be required to determine the role of DIO3 in bone.
46.7.3
Targeting thyroid hormone receptors
Studies in mutant mice have also revealed the roles of individual TR isoforms in the skeleton. Mutation or deletion of TRα causes severe skeletal abnormalities, including delayed endochondral ossification, impaired chondrocyte differentiation, defective growth plate development, and decreased bone mineralization. TRα 0/0 mice have complete deletion of all isoforms arising from the Thra locus and are systemically euthyroid with moderate skeletal abnormalities . Thus juveniles have transient growth delay with impaired endochondral and intramembranous ossification, whereas adults display increased bone volume and mineralization with osteosclerosis due to decreased osteoclastic bone resorption.
The key role of TRα1 in bone was further demonstrated in TRα1 PV mice that express a potent dominant-negative mutant receptor. The homozygous mutation is lethal, but TRα1 PV/− heterozygotes have mildly raised TSH and T3, but normal T4 levels. Despite their near normal thyroid status, TRα1 PV/+ mice have severely impaired skeletal development and grossly abnormal adult bone maintenance, displaying a much more severe phenotype than TRα 0/0 knockout mice and thus demonstrating an important deleterious role for the dominant-negative receptor in bone . TRα1 PV/+ mice that were also homozygous for a mutant nuclear corepressor NCoR1 that cannot interact with TR had a milder skeletal phenotype, indicating that interaction with NcoR1 is required for dominant-negative activity of the TRα1 PV mutant receptor . A similar developmental phenotype was seen in mice expressing a dominant-negative TRα1 L400R mutant in chondrocytes, indicating the important role of TRα1 in chondrocytes during skeletal development .
In contrast to TRα mutant mice in which thyroid status is near normal, TRβ −/− knockout mice have elevated circulating T4, T3, and TSH due to an impaired negative feedback response to thyroid hormones in the hypothalamus and pituitary . The elevated circulating thyroid hormones activate TRα signaling in bone, resulting in a phenotype of accelerated growth and endochondral ossification in juveniles and progressive bone loss and osteoporosis in adults that is similar to the effects of thyrotoxicosis . Furthermore, TRβ PV/PV mice that express dominant-negative TRβ mutant receptors have a more severe phenotype than TRβ −/− mice that consist of advanced endochondral and intramembranous ossification, craniosynostosis, short stature during development, and severe osteoporosis in adulthood . Together, these observations in TRβ mutant mice further demonstrate major roles for TRα1 in the developing and adult skeleton. Thus T3 stimulates anabolic actions mediated by TRα1 that promotes bone growth and mineralization during development but exerts catabolic actions of accelerated bone loss in adults that lead to osteoporosis.
46.7.4
Targeting the thyroid-stimulating hormone receptor
Tshr −/− knockout mice have congenital hypothyroidism and do not survive to adulthood unless supplemented with thyroid extract . Nevertheless, analysis of Tshr −/− mice revealed a 20% reduction in BMD, suggesting the hypothesis that TSH inhibits bone remodeling and turnover . Importantly, Tshr −/− mice received thyroid extract only after weaning and thus remained hypothyroid during the time of physiological peak thyroid hormone levels when growth velocity is at a maximum . Nevertheless, in hyperthyroid Tshr −/− mice treated with supraphysiological doses of thyroxine, bone loss was greater than observed in similarly treated wild-type control animals, suggesting further that TSH inhibits bone resorption in vivo and supporting the proposal that bone loss in hyperthyroidism may result from both elevated thyroid hormone concentrations and suppressed levels of TSH.
One study compared two strains of mutant mice, which both have grossly elevated circulating TSH levels and differ only by expressing a normal or nonfunctional TSHR. Thus Tshr hyt/hyt mice with congenital hypothyroidism harbor a loss-of-function mutation in Tshr , whereas Pax8 −/− mice lack the thyroid-specific transcription factor PAX8 essential for thyroid follicular cell development but express a normal fully functional TSHR. Both Tshr hyt/hyt and Pax8 −/− mice displayed similar skeletal abnormalities that are independent of TSHR signaling and consist of delayed endochondral ossification, growth retardation, reduced cortical bone volume, defective trabecular bone remodeling, and decreased bone mineralization. Furthermore, TRβ −/− and TRβ PV/PV mice each have disruption of the HPT axis with elevated circulating concentrations of both TSH and thyroid hormones. Both mutants display accelerated skeletal development and osteoporosis in adults that is consistent with elevated T3 signaling in bone but not with their high circulating TSH levels.
Overall, experimental studies in a series of mutant mice targeting all levels of the HPT axis and thyroid hormone signaling pathway are consistent with major physiological roles for T3, acting via TRα1, during skeletal development, and in adult bone. TSH also inhibits bone resorption via the TSHR in osteoclasts and suppression of TSH in thyrotoxicosis may thus compound the detrimental effects of thyroid hormone excess on the skeleton. Nevertheless, it is less clear whether TSH has important physiological effects during normal bone formation and maintenance.
46.8
Effects of thyroid signaling mutations on the human skeleton
A number of monogenic disorders affecting thyroid hormone action in humans have important consequences for the skeleton. These are classified as thyroid hormone cell membrane transporter defects, thyroid hormone metabolism defects, or thyroid hormone action defects .
46.8.1
Mutations affecting thyroid hormone transporters
Inactivating mutations in MCT8 cause Allan–Herndon–Dudley X-linked psychomotor retardation syndrome (OMIM 300523 ) with low T4 and elevated T3 levels. Affected individuals have severe developmental delay with ataxia, dystonia, and hypotonia, but the skeletal consequences have not been described in detail and are likely to be confounded by the neurological abnormalities of the condition .
46.8.2
Mutations affecting thyroid hormone metabolism
Selenocysteine (Sec) insertion sequence binding protein 2 (SBP2) mediates incorporation of the rare amino acid selenocysteine into the active site of the thyroid hormone deiodinases and is thus essential for conversion of T4 to T3. Affected individuals with mutations in SBP2 (OMIM 609698 ) have an elevated T4:T3 ratio and display transient delayed bone maturation, prepubertal growth retardation, and craniofacial abnormalities , demonstrating the importance of thyroid hormone metabolism and action for skeletal development in humans.
46.8.3
Mutations affecting thyroid hormone receptors
Resistance to thyroid hormone (RTH) β (OMIM 188570 ) is an autosomal-dominant condition caused by dominant-negative mutations of THRB , while RTHα results from dominant-negative mutations affecting THRA .
In RTHβ, mutant TRβ disrupts the negative feedback loop in the HPT axis, causing increased thyroid hormone levels with inappropriately normal or elevated TSH levels . Individuals have a variable and complex mixed phenotype of hyperthyroidism and hypothyroidism in different tissues, depending on (1) the degree of disruption to the HPT axis and consequent increase in circulating thyroid hormones that depends on the potency of the specific mutant receptor and (2) whether a particular tissue predominantly expresses TRα or TRβ. In the skeleton, affected individuals may develop craniofacial abnormalities, craniosynostosis, delayed or advanced bone age, short stature, and increased bone turnover causing osteoporosis and fracture, but a broad range of abnormalities have been observed even within affected kindreds .
Individuals affected by RTHα (OMIM 614450 ) have classical features of congenital hypothyroidism, but unlike RTHβ, there is no disruption to the normal feedback mechanisms of the HPT axis. TSH levels are normal, with a reduced T4:T3 ratio with normal/low T4 and normal/high T3 levels. Skeletal features result from severe disruption of thyroid hormone action in bone and include disproportionate short stature due to short lower limbs, delayed tooth eruption, delayed closure of fontanels, delayed bone age, epiphyseal dysgenesis, and a thickened calvarium. To date, 14 heterozygous frameshift, missense and nonsense mutations in THRA have been described that cause RTHα . The severity of the RTHα phenotype varies according to the type and location of the THRA mutations, with frameshift and nonsense mutations causing a more severe clinical syndrome than that seen in patients with milder missense mutations who may benefit from levothyroxine treatment .
46.8.4
Mutations affecting thyroid-stimulating hormone
Rare mutations have been described in TSHB that cause inactive TSH and congenital nongoitrous hypothyroidism (OMIM 275100 ). Normal BMD has been observed in two individuals aged 10 and 7 years with this condition who received thyroid hormone replacement from birth . Thus the lifelong absence of TSH did not affect bone mineralization during childhood.
Loss-of-function mutations in TSHR cause congenital nongoitrous hypothyroidism of varying severity (OMIM 275200 ) . Individuals with severe thyroid hypoplasia had delayed bone age and intramembranous ossification at birth but normal growth and skeletal development occurred following T4 replacement .
Nonautoimmune autosomal-dominant hyperthyroidism (OMIM 609152 ) is caused by rare gain-of-function mutations in TSHR and treated by surgical and radioiodine thyroid ablation followed by T4 replacement . At presentation, individuals display classical features of juvenile hyperthyroidism, including advanced bone age and craniosynostosis, but amelioration of the phenotype has been observed after treatment that normalizes systemic thyroid status . These conditions that affect TSHB and TSHR demonstrate that correction of thyroid hormone levels improves or prevents skeletal abnormalities, regardless of the levels and activity of TSH or the TSH receptor.
46.9
Skeletal effects of normal variation in thyroid status
46.9.1
Population genetic studies
Thyroid status is a highly heritable trait with complex inheritance. Normal individuals have a unique HPT axis set point that varies by less than 50% of the population reference range . Twin studies have supported these findings with heritability estimates of 23%–64% for circulating free T3 concentrations, 39%–65% for free T4 levels and 64%–65% for TSH . An initial meta-analysis of genome-wide association studies (GWASs) identified 20 loci independently associated with TSH levels and 6 loci independently associated with T4 levels . A recent meta-analysis that included over 72,000 individuals identified 109 variants associated with thyroid dysfunction and also led to the identification of a novel thyroid hormone transporter SLC17A4 and metabolizing enzyme AADAT . Genetic variation that affects tissue-specific thyroid hormone sensitivity is likely to affect variability in bone mineral density (BMD). BMD is also a highly heritable trait, and GWASs have identified over 500 loci to be independently associated with variability in BMD . Though the THRB locus was identified in a recent GWAS of BMD estimated by heel quantitative ultrasound in 426,824 UK Biobank participants , no other loci containing genes that regulate thyroid status have yet been associated with BMD or fracture susceptibility.
Candidate gene studies of thyroid signaling genes have yielded variable results when assessing the relationships between polymorphisms and changes in BMD. A large study of polymorphisms at the TSHR locus found no association with changes in femoral neck or lumbar spine BMD, and Mendelian randomization analysis found no evidence of a causal relationship between variation in TSH concentrations and BMD . However, the D727E polymorphism in TSHR had separately been associated with increased femoral neck BMD , but in apparent contradiction, the same polymorphism was found to have higher frequency in those with osteoporosis . Consistent with the UK Biobank GWAS , polymorphisms in THRB have been associated with trabecular but not cortical BMD at the hip , but variants in THRA have not been associated with BMD or fracture risk despite the autosomal-dominant skeletal dysplasia recently identified in patients with dominant-negative mutations of THRA . A polymorphism in DIO2 has been linked to reduced femoral neck BMD and increased levels of bone turnover markers, but the findings were not repeated in a larger study .
46.9.2
Epidemiological studies
Many observational studies have reported the consequences of altered thyroid function on BMD and fracture risk in population cohorts. Recently published studies have had larger cohort sizes, and some have included prospective follow-up. Larger cohorts reduce the potential for bias as many biological and environmental factors have large effects on BMD and fracture risk. Nonetheless, outcomes also depend on the composition of cohorts and may be confounded by inclusion of subjects with prior thyroid disease or varying combinations of pre- and postmenopausal female or male participants.
There have only been a few studies of the effects of thyroid status on bone turnover markers in euthyroid populations. In a study of 60 healthy postmenopausal women, high TSH levels were associated with low urinary deoxypyridinoline concentrations but not with serum procollagen type I C propeptide (PICP) levels . Two larger studies of postmenopausal women and men over 70 years of age , however, showed no association between TSH and serum osteocalcin, procollagen type 1 N-terminal propeptide (P1NP), beta-C-terminal telopeptide (CTX), and urinary N-terminal telopeptide (NTX).
The effect of variation of thyroid status within the normal reference range on BMD has recently been studied in a number of larger cohorts. Five cross-sectional studies of postmenopausal or elderly women with between 581 and 15,316 subjects found that low–normal TSH was associated with reduced hip or spine BMD . Three cross-sectional studies in men or both sexes (between 1017 and 1961 subjects) showed a consistent association between TSH and BMD in the forearm , lumbar spine , or hip . Only one recent study with 598 men and 719 women over 65 years of age showed no association between TSH levels and BMD . Other prospective and cross-sectional population studies (between 593 and 2957 subjects) have shown that high T4 is associated with reduced BMD or trabecular bone score , and a study of 4204 children showed that high–normal T4 is associated with lower BMD at 6 and 10 years of age . Although these studies have typically reported the association of either TSH or T4 with BMD, since both parameters are in a physiological inverse relationship, the results should be considered as an association with thyroid status rather than the effect of either TSH or T4 alone.
Low BMD is a major risk factor for fragility fractures, and so a number of studies have investigated associations between thyroid status and fracture risk directly. Recent prospective and cross-sectional studies of large female cohorts have shown that low TSH is associated with an increased risk of hip or vertebral fracture. Raised levels of T4 or T3 were also found to be associated with an increased risk of nonvertebral fracture by 20% and 33%, respectively . In one study of males aged over 65 years, low–normal TSH was associated with an increased risk of hip fracture . However, other population studies in postmenopausal or elderly women and/or men showed no significant association between TSH and fracture risk , including one large study of 16,610 women and 8595 men over 40 years of age . Despite these inconsistent data, a meta-analysis of individual participant data from 13 prospective studies of fracture risk in euthyroid individuals showed that low–normal TSH and high–normal T4 are associated with an increased risk of hip fracture . Importantly, this meta-analysis of individual participant data limited confounding due to study heterogeneity and led to a clear and robust conclusion.
In summary, these studies show that thyroid status in the upper–normal range with higher T4 and/or lower TSH is associated with decreased BMD and an increased risk of fracture.
46.10
Skeletal consequences of hypothyroidism
46.10.1
Hypothyroidism in children
Congenital hypothyroidism is the most common congenital endocrine disorder with an incidence of approximately 1:2000 to 1:4000 newborns and causes severe effects on growth with epiphyseal dysgenesis, delayed bone age, growth arrest, and short stature . This complex skeletal dysplasia includes hip subluxation, immature vertebral bodies, epiphyseal dysgenesis, absent secondary ossification centers in the femoral heads, delayed closure of the fontanelles, and a flattened nasal bridge . These features are remarkably similar to the phenotype observed in patients with RTHα, clearly demonstrating the role of TRα in the human skeleton. If congenital hypothyroidism is treated early, thyroxine replacement induces rapid catch-up growth with potential to achieve predicted adult height and BMD , though in one report adult BMD remained low despite treatment from the neonatal period .
Children with juvenile acquired hypothyroidism similarly display growth arrest and delayed bone maturation, but thyroxine treatment induces catch-up growth . Nonetheless, these individuals may fail to achieve the final predicted height, depending on the duration of thyroid hormone deficiency prior to initiation of treatment. There are no clinical data on the long-term consequences to the adult skeleton in individuals treated during childhood and adolescence for juvenile hypothyroidism.
46.10.2
Hypothyroidism in adults
In newly diagnosed patients with severe hypothyroidism, the bone resorption markers urinary pyridinoline and deoxypyridinoline are decreased . Histomorphometric analysis of bone biopsies of patients presenting with hypothyroidism has shown that bone turnover is also decreased with a net increase in bone formation . However, in these analyses, there was no significant increase in trabecular bone volume because structural consequences of low bone turnover require a long period of time to become evident. Consequently, BMD has also been found to be normal in newly diagnosed patients with hypothyroidism
Patients treated for hypothyroidism have been shown to have an increased risk of fracture . Nevertheless, fracture risk was proportional to the cumulative time and degree of thyroxine overreplacement, suggesting that bone loss and fracture susceptibility in hypothyroidism does not result from the disease but rather from the consequences of its treatment .
46.10.3
Subclinical hypothyroidism
Subclinical hypothyroidism is defined as a raised TSH in the presence of circulating thyroid hormone levels within the normal reference range, and it most commonly results from autoimmune thyroiditis. It has a high population prevalence, affecting 3% of men and 8% of women (10% of women over 55 years of age) . Most patients have no symptoms, though some questionnaire-based studies have shown an increased prevalence of tiredness and other nonspecific neurocognitive symptoms . Recent guidelines support treatment of selected individuals with subclinical hypothyroidism with thyroxine , although elderly populations in whom the age-specific reference range for TSH is increased are the least likely to benefit from treatment, and they are also the most likely to suffer unwanted side effects affecting the heart and cardiovascular system .
In large individual participant data meta-analyses, subclinical hypothyroidism was neither associated with a change in BMD nor with an increased risk of fracture . Overall, subclinical hypothyroidism has no significant effect on BMD or fracture risk, and there is no evidence of benefit to the skeleton from intervention. Importantly, thyroid hormone replacement stimulates bone turnover and may result in a detrimental reduction in BMD .
46.11
Skeletal consequences of hyperthyroidism
46.11.1
Hyperthyroidism in children
Thyrotoxicosis is rare in children, and Graves’ disease is the most common cause. In juvenile thyrotoxicosis, skeletal development is accelerated resulting in early cessation of growth and short stature due to premature fusion of the growth plates. In very young children, premature closure of the cranial sutures can cause craniosynostosis, resulting in cognitive impairment . Even in the developing skeleton, increased thyroid hormones can cause reduced BMD and decreased cortical bone density , though BMD returns to its predicted values after 1–2 years of treatment with antithyroid medication .
46.11.2
Hyperthyroidism in adults
Histomorphometry data from bone biopsies in patients with active thyrotoxicosis show increased bone turnover with a net increase in bone resorption . In thyrotoxicosis, bone turnover markers are increased, including both resorption markers such as urinary pyridinoline and deoxypyridinoline, and formation markers such as alkaline phosphatase and osteocalcin . Severe osteoporosis due to uncontrolled thyrotoxicosis is now rare because of prompt treatment with antithyroid medications or definitive treatment with thyroidectomy or radioactive iodine in refractory cases. Nonetheless, studies have revealed that BMD is reduced at the time of diagnosis and that fracture risk is increased . Moreover, the increase in fracture risk is proportionate to the duration of hyperthyroidism prior to treatment and persists for up to 5 years after treatment .
46.11.3
Subclinical hyperthyroidism
Subclinical hyperthyroidism, defined as normal T4 and T3 levels in the presence of suppressed or undetectable circulating TSH, is less common than subclinical hypothyroidism and has a prevalence of 1% and 1.5% in men and women over 60 years of age respectively . Though patients with subclinical hyperthyroidism are typically asymptomatic, there are significant long-term health consequences. Individuals with a TSH concentration below 0.1 mIU/L have increased all-cause mortality, coronary heart disease-related deaths, and risk of atrial fibrillation .
In patients with subclinical hyperthyroidism, the resorption markers urinary deoxypyridinoline and pyridinoline, and formation markers osteocalcin, alkaline phosphatase, and procollagen I C-terminal extension propeptide may be elevated or normal . Large meta-analyses have demonstrated that BMD is decreased and that fracture risk is increased with the largest effects in postmenopausal women . One meta-analysis of five large cohort studies found an increased risk of fracture in elderly subjects only . In a sub-analysis of the Fracture Intervention Trial cohort of 15,316 postmenopausal women, subjects with TSH <0.5 mIU/L had an increased risk of vertebral fracture, although individuals with overt thyrotoxicosis were not formally excluded . Only one recent meta-analysis failed to demonstrate a significant association between subclinical hyperthyroidism and fracture risk, but in this study, a broad definition of subclinical hyperthyroidism was adopted that included individuals with TSH only partially suppressed to <0.45 mIU/L . One prospective cohort study showed an association with hip fracture in men (increased hazard ratio of 3.27) but no clear association in women; however, only 47 out of 3646 study subjects were in the higher risk category of individuals with complete suppression of TSH .
Overall, the evidence from large individual participant data meta-analyses demonstrates that subclinical hyperthyroidism is associated with detrimental consequences for BMD and fracture risk . The association with osteoporosis in these studies is consistent with the population effects of moderate increases in thyroid function within the normal range. In postmenopausal women the risk of subclinical hyperthyroidism causing osteoporosis is higher than in premenopausal women or men and is increased in proportion to the severity of TSH suppression.
46.11.4
Patients treated for hyperthyroidism
Hyperthyroidism results in a state of high bone turnover with a net increase in bone resorption that leads to osteoporosis. Various studies have examined the effects of treatment for thyrotoxicosis on the skeleton. Following treatment with antithyroid drugs, the elevated bone resorption and formation markers return to normal within 2 months . The low BMD at the time of diagnosis similarly returns to normal within 1–4 years , though if thyrotoxicosis is treated by thyroidectomy, improvement in BMD may occur as rapidly as 6 months following surgery . Despite the rapid effects of antithyroid medication on bone turnover and BMD, in a large case–control fracture risk study comprising half a million subjects, the increased relative risk of fracture persisted to 5 years after initiation of treatment and may result from inadequate treatment or noncompliance .
46.11.5
Thyroid-stimulating hormone suppression for treatment of differentiated thyroid cancer
Differentiated thyroid cancer comprises both papillary and follicular thyroid cancer, and treatment after total thyroidectomy and radioiodine ablation may require long-term suppression of serum TSH with thyroxine . Similar to other causes of subclinical hyperthyroidism, this exogenous cause carries the risk of osteoporosis and fragility fracture, in addition to an increased risk of atrial fibrillation and cardiovascular mortality .
Small cohorts of patients receiving suppressive doses of T4 have been studied in detail and were found to have increased levels of bone resorption and formation markers . Most studies of BMD have shown TSH suppression therapy to be associated with reduced BMD at the lumbar spine, femur or radius in postmenopausal women proportionate to the duration of TSH suppression . Despite this, TSH suppression therapy was not associated with change in BMD in male patients .
As differentiated thyroid cancer frequently has a favorable prognosis, international guidelines now recommend dynamic risk stratification of patients with the aim to limit the duration and severity of required TSH suppression . Patients with low or intermediate risk of recurrent disease should have moderate TSH suppression to <2.0 or 0.1–0.5 mIU/L, respectively . The highest risk patients should have target TSH levels of <0.1 mIU/L rather than complete suppression of TSH to undetectable levels. This is because complete suppression of TSH is associated with the highest risk of osteoporotic fractures, with no additional reduction in risk of tumor recurrence . Since suppressive doses of thyroxine have a greater effect on the skeleton in postmenopausal women , monitoring of BMD has been recommended in this group of patients .
46.12
Conclusion
- •
The developing and adult skeleton is exquisitely sensitive to thyroid status. T3 exerts anabolic responses during skeletal growth and catabolic effects on adult bone.
- •
Circulating thyroid hormones and TSH are precisely regulated by the HPT axis that constitutes a classical endocrine negative feedback loop.
- •
Epidemiological studies show that thyroid status even in the upper–normal range is associated with decreased BMD and an increased fracture risk.
- •
Hyperthyroidism results in reduced BMD and an increased incidence of fracture that normalizes following treatment.
- •
In postmenopausal women, subclinical hyperthyroidism increases bone fragility proportionately with the degree of TSH suppression.
- •
Overreplacement of thyroxine in patients with hypothyroidism and TSH suppression for treatment of differentiated thyroid cancer is associated with an increased risk of fracture.
46.13
Author short biographies
Dr. Bernard Freudenthal trained in medicine at Cambridge University and University College London and is a specialist trainee in Endocrinology and Diabetes. He is currently undertaking PhD studies as an Academic Clinical Fellow funded by a highly prestigious Medical Research Council Clinical Research Training Fellowship.
Dr. Laura Watts trained in medicine at Oxford University and is a specialist trainee in Rheumatology. She is currently undertaking PhD studies funded as an Academic Clinical Fellow by a highly prestigious Wellcome Trust Imperial College 4i Clinical Research Fellowship.
Professor JH Duncan Bassett trained in medicine at Cambridge and Oxford Universities and is currently Professor of Endocrinology at Imperial College London and a consultant physician in metabolic bone disease at the Hammersmith Hospital in London.
Professor Graham R Williams trained in medicine at St. Thomas’s Hospital Medical School, University of London and is currently Professor of Endocrinology at Imperial College London and a consultant physician in thyroid disease and endocrinology at the Hammersmith Hospital in London.
Acknowledgments
The authors gratefully acknowledge the generous funding as follows. BF is supported by a Medical Research Council Clinical Research Training Fellowship (MR/P018718/1), LW by a Wellcome Trust Imperial College 4i Clinical Research Fellowship, and JHDB and GRW are funded by a Wellcome Trust Strategic Award (101123), Wellcome Trust Joint Investigator Award (110140 and 110141), European Commission Horizon 2020 Grant (666869, THYRAGE), and a Medical Research Council Research Grant (MR/N01121X/1).
References
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


