Since the last review in 2007 of thrombotic thrombocytopenic purpura (TTP) and microangiopathic hemolytic anemia in the Clinics , further understanding of the nature of TTP and atypical hemolytic uremic syndrome (aHUS) has led to increasing use of rituximab in the treatment of TTP and the approval in 2011 of eculizumab for the treatment of aHUS. With this new armamentarium, distinction of aHUS from TTP has become more critical than ever. This article updates the new knowledge, highlights the difference between aHUS and TTP, and presents a scheme for their diagnosis and management.
Key points
- •
Both thrombotic thrombocytopenic purpura (TTP) and atypical hemolytic uremic syndrome (aHUS) are chronic diseases that often present with thrombocytopenia and microangiopathic hemolysis (MAHA), yet they have entirely different pathology and pathogenesis and require different therapeutic approaches.
- •
Patients presenting with thrombocytopenia and MAHA are started on plasma exchange therapy for presumed TTP unless history and laboratory test results clearly indicate that patients have one of the disorders that do not require plasma therapy.
- •
Plasma exchange is continued for acquired TTP until clinical remission. The treatment may be switched to plasma infusion for hereditary TTP unless the patient has renal failure. Depending on its course, acquired TTP may require rituximab treatment to prevent relapses.
- •
Plasma exchange is switched to eculizumab for patients without severe ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 motif, member 13) deficiency and considered to have aHUS.
- •
Both TTP and aHUS are chronic diseases that require long-term monitoring and management.
A major challenge in the management of patients presenting with thrombocytopenia and microangiopathic hemolytic anemia (MAHA) is making a distinction between TTP and aHUS. The discovery of ADAMTS13 and its deficiency in TTP has provided a pathogenetic definition of TTP. Nevertheless, some investigators continue to view aHUS as a subtype of TTP without severe ADAMTS13 deficiency. Under such schemes, aHUS in adult patients is treated indiscriminately from TTP.
In reality, TTP and aHUS not only differ in pathology, pathogenesis, pathophysiology, and prognosis but also require different therapeutic management. The similarity between TTP and aHUS in causing thrombocytopenia and microangiopathic hemolysis is an epiphenomenon of microvascular stenosis resulting from entirely different mechanisms.
Fragmentation of the red blood cells occurs in 2 types of clinical conditions: vascular devices, such as prosthetic heart valves, ventricular assist devices, and extracorporeal oxygenator, and microvascular stenosis. These 2 conditions share a common feature of abnormal intravascular shear stress that is sufficient to cause fragmentation of the red blood cells ( Fig. 1 ). In the absence of vascular devices, fragmentation of the red blood cells signifies stenosis in the arterioles and capillaries.
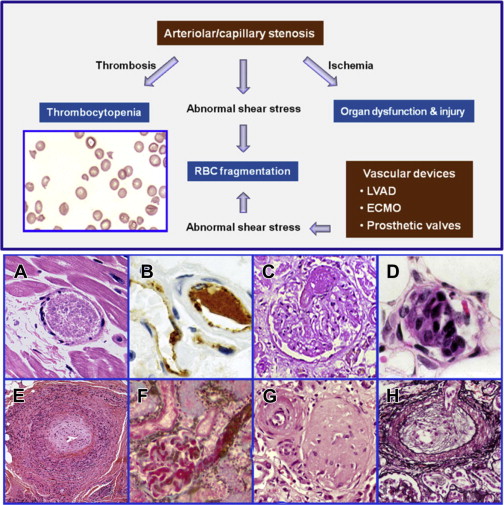
Pathologically at least 5 different types of arteriolar stenosis are observed (see Fig. 1 ): (1) von Willebrand factor (VWF) platelet thrombosis, typically observed in patients with TTP due to severe ADAMTS13 deficiency; (2) platelet fibrin thrombosis, as exemplified in patients with disseminated intravascular coagulopathy (DIC); (3) tumor cell invasion of the microvasculature in patients with metastatic neoplasm; (4) microvascular vasculitis complicating autoimmune or certain infectious disorders; and (5) thrombotic microangiopathy, as observed in patients with the typical shiga toxin–associated hemolytic uremic syndrome after certain Escherichia coli infection or aHUS due to defective regulation of the alternative complement pathway.
In thrombotic microangiopathy, endothelial changes are prominent. Endothelial cell swelling or disruption, accompanied by intimal expansion and cellular proliferation, may cause microvascular stenosis or occlusion with or without thrombosis. In addition, edema of the brain and other organs and fluid accumulation in cavitary spaces due to abnormal vascular permeability may contribute to organ dysfunction in patients with thrombotic microangiopathy. In TTP, tissue injury results from ischemia of microvascular thrombosis; the endothelium and vessel wall structures are intact and complications of abnormal vascular permeability do not occur.
The various types of microvascular stenosis, in particular types 1 to 4, are often incorrectly referred to as thrombotic microangiopathy without distinction. Furthermore, thrombocytopenia and microangiopathic hemolysis are often equated in practice with thrombotic microangiopathy, ignoring other types of pathology causing microvascular stenosis. Both practices obscure the important differences among the various types of pathology and contribute to the unfounded view of TTP and aHUS as one disease entity.
Pathogenesis and pathophysiology of TTP and aHUS
The pathogenesis of TTP and aHUS are different. TTP results from severe ADAMTS13 deficiency due to genetic mutations or, more commonly, autoimmune inhibitors, whereas aHUS results from defective regulation of the complement system. Although a recent report describes the activation of the complement system in TTP, presumably by the ADAMTS13-inhibitor complexes, there is no evidence that complement dysregulation contributes to the development of TTP.
ADAMTS13 is a major determinant preventing VWF platelet aggregation in the normal circulation. Inflammation, infection, surgery, or pregnancy may decrease the plasma ADAMTS13 activity level by suppressing the biosynthesis of ADAMTS13 or possibly enhancing its inactivation. Although inflammation or pregnancy-mediated decrease in ADAMTS13 per se is insufficient to induce microvascular thrombosis, it may lead to disease exacerbation in patients with preexisting TTP.
In the presence of severe ADAMTS13 deficiency, the propensity of VWF and platelet to form aggregates is affected by multiple other factors, including the platelet count, the conformational responsiveness of VWF to activation by shear stress, and, most importantly, the shear stress profile in the microcirculation.
In vitro, the critical shear stress required for VWF platelet aggregation is near the high end of physiological range (50–90 dyn/cm 2 ). Therefore, in patients with shear stress profiles that are not sufficient to activate the VWF in their microcirculation, VWF platelet aggregation may not occur even when ADAMTS13 deficiency is severe. This may explain why some patients with severe ADAMTS13 deficiency are asymptomatic and have normal platelet counts. Any condition, however, such as fever, infection, or surgery, may trigger microvascular thrombosis, causing symptomatic complications in such patients.
Animal Models of ADAMTS13 Deficiency and Modifiers of VWF Platelet Aggregation
Two types of animal models have been developed to investigate the role of the protease in preventing microvascular thrombosis: mice with inactivated ADAMTS13 gene and baboons given an inhibitory ADAMTS13 antibody.
Inactivation of the ADAMTS13 gene has produced intriguing results in mice. It produces spontaneous microvascular thrombosis in some but not other strains. Shiga toxins may induce microvascular thrombosis in the susceptible strains by inducing the release of VWF from endothelial cells. In the resistant strains, infusion of large amount of human VWF leads to development of microvascular thrombosis, suggesting that the mouse VWF platelet axis is less responsive to ADAMTS13 deficiency.
Infusion of an inactivating monoclonal antibody of ADAMTS13 in baboons leads to the development of microvascular thrombosis, confirming the antithrombotic role of ADAMTS13 in normal circulation.
Pathogenesis and Pathophysiology of aHUS
aHUS results from defects in the regulation of the complement system ( Fig. 2 ). Three types of molecular defects or variants are detected in aHUS patients: inactivating mutations of complement factor H (CFH; 25%), membrane cofactor protein (MCP, ie, CD46), complement factor I (CFI), or thrombomodulin (THBD) (approximately 5%–10% each); gain-of-function mutations of complement factor B (CFB) or C3 approximately 5%–10%); and autoantibodies of CFH (approximately 5%–10%). Additionally, certain common variants of CFH, CFH-related protein (CFHR) 2 and other regulators; or their combinations (haplotypes and complotypes) may increase the risk of aHUS. Because the list of affected molecules remains incomplete, negative genetic or antibody test results do not exclude the diagnosis of aHUS.
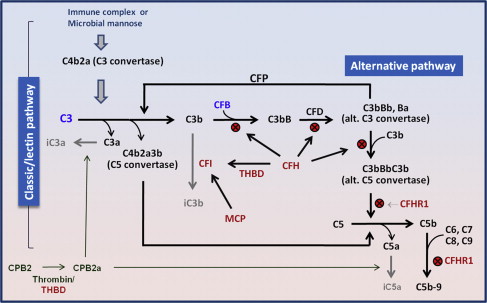
Defective regulation of the complement activation leads to excess generation of cytotoxic C5b-9 (ie, membrane attack complex) and anaphylatoxins C3a and C5a. Membrane attack complex causes cytotoxicity of endothelial cells, leading to endothelial swelling or disruption and intimal swelling and cellular proliferation. Endothelial disruption exposes the prothrombotic components in the subendothelial space, leading to activation of the coagulation system and fibrin deposition ( Fig. 3 ).
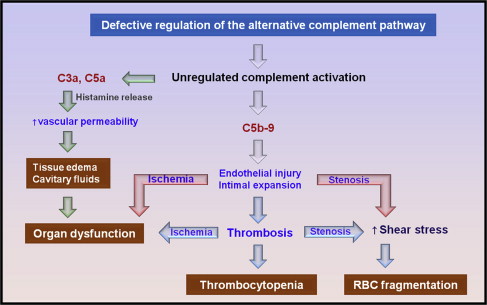
In aHUS, microvascular stenosis may result not only from thrombosis but also directly from endothelial swelling and subendothelial expansion. Furthermore, abnormal vascular permeability mediated by C3a and C5a may cause interstitial edema of the brain and other vital organs and cavitary fluid accumulation. Therefore, mental changes, seizures, cardiac dysfunction or arrest, pericardial effusion, chest pain, dyspnea, pleural effusion, pulmonary infiltrates, abdominal pain, nausea, vomiting, diarrhea, pancreatitis, ascites, renal failure, and anasarca may occur in aHUS without concurrent worsening of MAHA or thrombocytopenia.
Pathogenesis and pathophysiology of TTP and aHUS
The pathogenesis of TTP and aHUS are different. TTP results from severe ADAMTS13 deficiency due to genetic mutations or, more commonly, autoimmune inhibitors, whereas aHUS results from defective regulation of the complement system. Although a recent report describes the activation of the complement system in TTP, presumably by the ADAMTS13-inhibitor complexes, there is no evidence that complement dysregulation contributes to the development of TTP.
ADAMTS13 is a major determinant preventing VWF platelet aggregation in the normal circulation. Inflammation, infection, surgery, or pregnancy may decrease the plasma ADAMTS13 activity level by suppressing the biosynthesis of ADAMTS13 or possibly enhancing its inactivation. Although inflammation or pregnancy-mediated decrease in ADAMTS13 per se is insufficient to induce microvascular thrombosis, it may lead to disease exacerbation in patients with preexisting TTP.
In the presence of severe ADAMTS13 deficiency, the propensity of VWF and platelet to form aggregates is affected by multiple other factors, including the platelet count, the conformational responsiveness of VWF to activation by shear stress, and, most importantly, the shear stress profile in the microcirculation.
In vitro, the critical shear stress required for VWF platelet aggregation is near the high end of physiological range (50–90 dyn/cm 2 ). Therefore, in patients with shear stress profiles that are not sufficient to activate the VWF in their microcirculation, VWF platelet aggregation may not occur even when ADAMTS13 deficiency is severe. This may explain why some patients with severe ADAMTS13 deficiency are asymptomatic and have normal platelet counts. Any condition, however, such as fever, infection, or surgery, may trigger microvascular thrombosis, causing symptomatic complications in such patients.
Animal Models of ADAMTS13 Deficiency and Modifiers of VWF Platelet Aggregation
Two types of animal models have been developed to investigate the role of the protease in preventing microvascular thrombosis: mice with inactivated ADAMTS13 gene and baboons given an inhibitory ADAMTS13 antibody.
Inactivation of the ADAMTS13 gene has produced intriguing results in mice. It produces spontaneous microvascular thrombosis in some but not other strains. Shiga toxins may induce microvascular thrombosis in the susceptible strains by inducing the release of VWF from endothelial cells. In the resistant strains, infusion of large amount of human VWF leads to development of microvascular thrombosis, suggesting that the mouse VWF platelet axis is less responsive to ADAMTS13 deficiency.
Infusion of an inactivating monoclonal antibody of ADAMTS13 in baboons leads to the development of microvascular thrombosis, confirming the antithrombotic role of ADAMTS13 in normal circulation.
Pathogenesis and Pathophysiology of aHUS
aHUS results from defects in the regulation of the complement system ( Fig. 2 ). Three types of molecular defects or variants are detected in aHUS patients: inactivating mutations of complement factor H (CFH; 25%), membrane cofactor protein (MCP, ie, CD46), complement factor I (CFI), or thrombomodulin (THBD) (approximately 5%–10% each); gain-of-function mutations of complement factor B (CFB) or C3 approximately 5%–10%); and autoantibodies of CFH (approximately 5%–10%). Additionally, certain common variants of CFH, CFH-related protein (CFHR) 2 and other regulators; or their combinations (haplotypes and complotypes) may increase the risk of aHUS. Because the list of affected molecules remains incomplete, negative genetic or antibody test results do not exclude the diagnosis of aHUS.
Defective regulation of the complement activation leads to excess generation of cytotoxic C5b-9 (ie, membrane attack complex) and anaphylatoxins C3a and C5a. Membrane attack complex causes cytotoxicity of endothelial cells, leading to endothelial swelling or disruption and intimal swelling and cellular proliferation. Endothelial disruption exposes the prothrombotic components in the subendothelial space, leading to activation of the coagulation system and fibrin deposition ( Fig. 3 ).
In aHUS, microvascular stenosis may result not only from thrombosis but also directly from endothelial swelling and subendothelial expansion. Furthermore, abnormal vascular permeability mediated by C3a and C5a may cause interstitial edema of the brain and other vital organs and cavitary fluid accumulation. Therefore, mental changes, seizures, cardiac dysfunction or arrest, pericardial effusion, chest pain, dyspnea, pleural effusion, pulmonary infiltrates, abdominal pain, nausea, vomiting, diarrhea, pancreatitis, ascites, renal failure, and anasarca may occur in aHUS without concurrent worsening of MAHA or thrombocytopenia.
A pathogenetic classification of MAHA
Microangiopathic hemolysis may be classified according to its pathogenetic mechanisms or pathology ( Table 1 ). This scheme is anchored on group I of TTP with severe ADAMTS13 deficiency due to autoimmune inhibitors or genetic mutations, and group II of aHUS with defective complement regulation due to genetic mutations or autoantibodies of the activators or regulators of the alternative complement pathway. Included in either group I or II are comorbid conditions, such as infection, inflammation, surgery, trauma, pregnancy, intravenous contrast agents, and pancreatitis, that may trigger acute presentation in patients with preexisting TTP or aHUS, either by promoting the VWF platelet interaction or by activating the complement system (group IB or IIB). Pregnancy is considered group II comorbidity for aHUS because most pregnant women presenting with hemolytic-uremic syndrome (HUS) have molecular defects in the regulation of the alternative complement pathway.
|
|
|
|
Related posts:
 Harnessing the Platelet Signaling Network to Produce an Optimal Hemostatic Response
Harnessing the Platelet Signaling Network to Produce an Optimal Hemostatic Response
 Platelet Transfusion Therapy
Diagnosis and Management of Heparin-Induced Thrombocytopenia
Platelet Transfusion Therapy
Diagnosis and Management of Heparin-Induced Thrombocytopenia
 Harnessing the Platelet Signaling Network to Produce an Optimal Hemostatic Response
Drug-induced Immune Thrombocytopenia
Diagnosis and Management of Heparin-Induced Thrombocytopenia
Harnessing the Platelet Signaling Network to Produce an Optimal Hemostatic Response
Drug-induced Immune Thrombocytopenia
Diagnosis and Management of Heparin-Induced Thrombocytopenia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


