Thrombotic Complications of Sickle Cell Disease
Kenneth I. Ataga
Elliott P. Vichinsky
In addition to its well described vaso-occlusive and hemolytic complications, patients with sickle cell disease (SCD) are at risk for a variety of thrombotic complications. Patients with SCD have an increased risk of stroke, pulmonary hypertension, avascular necrosis of large joints, pregnancy-related complications, and leg ulcers, all of which may be due, at least in part, to thrombotic vascular occlusion.1 A retrospective study based on reported discharge diagnoses showed that SCD patients were more likely to be diagnosed with pulmonary embolism compared with race-matched controls without SCD (0.44% vs. 0.12%), although interestingly, the prevalence of deep vein thrombosis was similar between the two groups.2 SCD also appears to be a significant risk factor for pregnancy-related venous thromboembolism, with an odds ratio of 6.7 (95% confidence interval [CI]: 4.4 to 10.1).3, 4 Finally, although distinct from SCD in its clinical manifestations, individuals with the sickle cell trait appear to be at increased risk of venous thromboembolism compared to race-matched controls, with an odds ratio of about two.5, 6 Sickle cell trait appears to be associated with pulmonary embolism, rather than deep vein thrombosis,5 analogous to the situation in patients with SCD.2
▪ THE RED BLOOD CELL AND HEMOGLOBIN S POLYMERIZATION
Sickle hemoglobin (HbS) occurs when the normal β6 glutamic acid residue is replaced by valine (GAG to GTG mutation at codon β6). The polymerization of HbS (α2β2S) following its deoxygenation is the primary event in the pathophysiology of SCD and results in damage to red blood cells (RBCs), tissues, and organs.7 The rate and extent of HbS polymer formation depend on the degree of deoxygenation of HbS, the intraerythrocytic HbS concentration, and the intracellular concentration of fetal hemoglobin (HbF).7 The lag period required for the formation of HbS polymer is referred to as the delay time. As the range of transit times in the microcirculation is short relative to the range of delay times of sickle RBCs, polymers do not form in most of the cells.8 HbF inhibits the polymerization of HbS, primarily owing to the glutamine residue at codon γ87, which prevents a critical lateral contact in the double strand of the sickle fiber.7
Patients with SCD exhibit an unusually broad density distribution of sickle RBCs due mainly to the high number of reticulocytes, with a relatively low intracellular hemoglobin concentration, and the presence of a high number of very dense cells. Dense cells occur due to enhanced cellular dehydration following polymerization-induced damage to the cell membrane.7 As HbS polymerization is strongly dependent on the intracellular hemoglobin concentration,9 dense sickle RBCs are more likely to polymerize and contribute to the hemolytic and vaso-occlusive complications of SCD than less dehydrated cells.
▪ LEUKOCYTES
Leukocytes play an important role in the pathogenesis of SCD. Increased white blood cell count is a risk for early death,10 acute chest syndrome,11 and hemorrhagic stroke12 in patients with sickle cell anemia. In addition, severe episodes of vaso-occlusive crisis and acute chest syndrome have occurred following the administration of granulocyte-colony stimulating factor (G-CSF) to patients with SCD in their steady state.13, 14 Leukocytes interact with sickle RBC and vascular endothelium and are stimulated to release injurious cytokines.15, 16 In addition to the adhesion to vascular endothelium, leukocytes interact with erythrocytes and platelets to form cell aggregates, stabilized via CD36-TSP, CD31-CD31, and CD62L-CD162 bonds.17, 18 Using intravital microscopy in mice expressing human HbS, sickle RBCs were observed to bind to adherent leukocytes in inflamed venules, producing vaso-occlusion of cremasteric venules.17 However, both the adhesion of leukocytes to endothelium and the interaction of leukocytes with sickle RBCs were substantially reduced in sickle mice that were deficient in P- and E-selectins, suggesting that adhesion to endothelial selectins may enhance the ability of adherent leukocytes to bind circulating sickle RBCs.
Monocytes are activated in sickle cell anemia and can enhance vaso-occlusion through an endothelial inflammatory response promoted by the nuclear factor-κB (NFκB)-mediated upregulation of adhesion molecules and tissue factor (TF).19 The activation of endothelial cells by sickle monocytes appears to be mediated by tumor necrosis factor-α and interleukin-1β, both markers of monocyte activation. The activated endothelial cells increase their expression of ligands for adhesion molecules on leukocytes and RBCs, thereby promoting vaso-occlusion.
▪ ENDOTHELIUM
As the potential for sickle RBCs to initiate a vaso-occlusive event is dependent on whether the rate of polymer formation is within the range of the capillary transit time,20 any factor that slows the transit of these cells through the microcirculation could be expected to have an effect on the pathogenesis of vaso-occlusion. Multiple studies demonstrate that sickle RBCs attach more readily to cultured endothelial cells than do normal RBCs.21, 22, 23, 24 Indeed, the degree of adherence of sickle RBCs to vascular endothelium strongly correlates with disease severity.21
In addition to WBC adhesion to endothelium, adhesion reactions are mediated by interactions between receptors on sickle RBCs and endothelial cells or subendothelial matrix proteins. The plasma ligand, thrombospondin (TSP) provides a bridge between the RBC receptor CD36 and several constitutively expressed endothelial receptors,25, 26 including the vitronectin receptor (αvβ3), the transmembrane glycoprotein CD36, and endothelial cell surface heparan sulfate proteoglycans. Sickle RBCs also interact with immobilized TSP via the integrin-associated protein, CD47.27 Interactions occur between the integrin complex, α4β1 (VLA-4), expressed on reticulocytes, and both endothelial vascular-cell adhesion molecule 1 (VCAM-1)28, 29, 30, 31 (typically expressed on the surface of endothelial cells following activation by inflammatory cytokines and hypoxia) and fibronectin.32 High molecular weight multimers of von Willebrand factor (vWF)33, 34 promote RBC adhesion to endothelial vitronectin receptor, αvβ3 and the GPIb-IX-V complex. Interactions also occur between sickle RBC and subendothelial immobilized matrix proteins, including laminin via the erythrocyte basal cell adhesion molecule-Lutheran protein receptor, the protein that carries Lutheran blood-group antigens,35 as well as TSP, vWF, and fibronectin, proteins that are also present in plasma in a soluble form.36 Nonreceptor-mediated adhesive mechanisms include a role for RBC sulfated glycolipids and phosphatidylserine (PS).37, 38, 39
▪ INFLAMMATION
SCD is often referred to as a chronic inflammatory state due to the presence of a chronic elevation in leukocyte counts, abnormal activation of cellular elements including neutrophils and monocytes,40, 41 circulating endothelial cells,42 and platelets43, 44 as well as activation of the coagulation system.1 In addition, patients exhibit elevated levels of inflammatory mediators (such as interleukin-6, tumor necrosis factor α, interleukin-1, placenta growth factor [PlGF]), acute phase reactants (such as C-reactive protein, secretory phospholipase A2, and G-CSF), and markers of endothelial cell injury (such as soluble VCAM)40, 45, 46 even in the non-crisis, “steady state.” The inflammatory biology in SCD patients may result from infection, as well as iron replete (or overloaded) states, sickle RBC adhesion to endothelium, and the reperfusion-injury physiology observed in these patients.40
▪ COAGULATION ACTIVATION
As nearly every component of hemostasis, including platelet function, and the procoagulant, anticoagulant, and fibrinolytic systems, is altered in the direction of a procoagulant phenotype, SCD is frequently referred to as a “hypercoagulable state1 (FIGURE 122.1).” Patients exhibit a loss of normal membrane phospholipid asymmetry in a subpopulation of RBCs, occurring in mature cells, RNA-containing reticulocytes, and transferrin receptor-positive “stress” erythrocytes.47, 48 PS is normally found in the inner monolayer of the plasma cell membrane, whereas choline-containing phospholipids such as phosphatidylcholine and sphingomyelin are located in the outer monolayer.49 RBC membrane phospholipid asymmetry is usually maintained by the action of an ATP-dependent aminophospholipid translocase (or flipase), that transports PS and phosphatidylethanolamine from the outer to the inner membrane surface.49 In addition, movement of all phospholipids in both directions is produced by scramblase, following activation by an influx of extracellular calcium, resulting in rapid PS exposure.50 The abnormal PS exposure in sickle RBCs may occur due to repeated cycles of sickling and unsickling linked to polymerization and depolymerization of HbS, which results in the production of terminal spicules or microvesicles with exposed PS.51 The abnormal exposure of PS may also result from reduced flipase activity in RBCs due to oxidative stress and sulfhydryl modification.49, 52, 53 Abnormal PS exposure functions as a recognition signal for cell removal during apoptosis of nucleated cells,54 as well as a docking site for enzymatic complexes involved in coagulation and anticoagulation pathways.50 The external exposure of PS alters the adhesive properties of sickle RBCs55 and has been reported to be involved in the hemostatic changes observed in SCD.56, 57 Several data show evidence for a relationship between RBC PS exposure and the coagulation activation in SCD. Plasma levels of prothrombin fragment 1.2 (F1.2) are associated with the number of circulating PS-positive RBCs.57 However, although the number of PS-positive sickle RBCs is significantly correlated with plasma F1.2, D-dimer, and plasmin-antiplasmin (PAP) complexes, no correlation is found between PS-positive platelets and any of these markers of coagulation activation,56 suggesting that sickle RBCs, and not platelets, are responsible for the coagulation activation observed in SCD. Finally, sickle RBCs containing high amounts of HbF are less likely to result in microparticle (MP) formation, PS exposure, and thrombin generation,57 suggesting a protective effect of HbF on membrane bilayer flip-flop and PS exposure.
Measures of hemolysis are associated with plasma markers of coagulation activation such as thrombin-antithrombin complexes (TAT), F1.2, and D-dimers in patients with SCD.45, 58 Heme, an inflammatory mediator and a product of intravascular hemolysis, has been reported to induce TF expression on the surface of both macrovascular and microvascular endothelial cells in a concentration-dependent manner.59 In addition, heme resulted in an upregulation of the expression of TF mRNA, TF protein, and TF procoagulant activity in endothelial cells in a time-dependent manner, effects that may be mediated, at least in part, by the transcription factor, NFκB. Heme-induced endothelial TF expression may provide a pathophysiologic link between hemolysis and the coagulation activation observed in patients with SCD.
The inflammatory state in SCD may also contribute to coagulation activation. Studies in transgenic SCD mice demonstrate that exposure of the mild sickle cell phenotype, NY1DD mice, to a hypoxic environment for 3 hours followed by a return to ambient air for 18 hours resulted in increased TF expression in the pulmonary veins. Thus, ischemia-reperfusion injury in patients with SCD may play a pathophysiologic role in the activation of coagulation.60 The endothelial TF expression in the NY1DD mouse appears to be highly dependent on NFκB (p50) in peripheral blood mononuclear cells but not in the vessel wall.61 Furthermore, a study of NY1DD animals bred to overexpress endothelial nitric oxide synthase (eNOS) or to have an eNOS knockout state demonstrated that eNOS modulates endothelial TF expression in vivo by downregulating it, suggesting that the biodeficiency of NO, characteristic of SCD patients, may increase the risk of coagulation activation.62
Finally, increased levels of soluble CD40 ligand, a tumor necrosis factor family member, may contribute to coagulation activation in SCD. Plasma from patients with sickle cell anemia
induced a significant increase in TF production in monocytic THP-1 cells relative to plasma from HbAA individuals or media alone.44 Furthermore, preincubation of the THP-1 cells with a function blocking anti-CD40 antibody prevented increased TF expression in five of eight individual cases, suggesting that the CD40:CD40L interaction is only partly responsible for the elevation in TF production.
induced a significant increase in TF production in monocytic THP-1 cells relative to plasma from HbAA individuals or media alone.44 Furthermore, preincubation of the THP-1 cells with a function blocking anti-CD40 antibody prevented increased TF expression in five of eight individual cases, suggesting that the CD40:CD40L interaction is only partly responsible for the elevation in TF production.
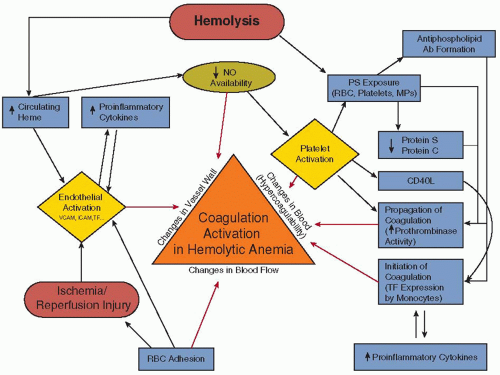 FIGURE 122.1 Schematic representation of pathophysiologic mechanisms (described or postulated) leading to activation of coagulation in SCD. Based on Virchow triad, the illustrated pathways contribute to activation of coagulation (and possibly eventual thrombosis) by one of three broad mechanisms, that is, (1) changes in the vessel wall; (2) changes in blood flow; and/or (3) changes in the composition of blood components (“hypercoagulability”). NO, nitric oxide; PS, phosphatidylserine; RBC, red blood cell; MPs, microparticles; TF, tissue factor; SCD, sickle cell disease. (Modified from Ataga KI, Key NS. Hypercoagulability in sickle cell disease: new approaches to an old problem. Hematology Am Soc Hematol Educ Program 2007:91-96.) |
▪ INCREASED TISSUE FACTOR EXPRESSION
The TF-FVIIa complex is the physiologic initiator of hemostasis.63 Patients with SCD exhibit abnormal expression of TF on circulating endothelial cells, and this TF expression is increased further during acute pain episodes.64 In addition, both TF antigen and TF procoagulant activity are elevated in the circulation of SCD patients when compared with normal controls,44, 65, 66 although no difference in whole blood TF procoagulant activity was observed in patients in the non-crisis, “steady state” compared to those with pain crises.65
While numerous plasma factors that could increase TF expression in hematopoietic and/or endothelial cells such as thrombin, tumor necrosis factor, interleukin-1, and endotoxin are elevated in SCD patients, multiple potential mechanisms for the increased TF expression in these patients have recently been described. As already described, these include hemolysis with increased levels of heme,59 ischemia-reperfusion injury,60 as well as increased plasma levels of soluble CD40 ligand.44
▪ CHANGES IN MARKERS OF THROMBIN GENERATION AND ANTICOAGULANT PROTEINS
Patients with SCD have increased plasma levels of markers of thrombin generation in the non-crisis, “steady state,” although there are conflicting data on the effect of acute pain episodes on the levels of these markers.67, 68 Plasma levels of F1.2 and TAT complexes, both markers of thrombin generation, are elevated in SCD patients in the noncrisis, steady state.67, 68 Plasma levels of D-dimers,66, 67, 68 PAP complexes,67 and fibrinopeptide A68 are also observed to be elevated in SCD patients in the non-crisis, “steady state.” The frequency of pain episodes correlates with the extent of fibrinolytic activity (assessed by D-dimer levels) in
the non-crisis, “steady state,” suggesting that D-dimer levels may predict the frequency of pain crises.67‘
the non-crisis, “steady state,” suggesting that D-dimer levels may predict the frequency of pain crises.67‘
The levels of natural anticoagulant proteins, protein C and protein S, are also decreased68, 69 in the non-crisis, “steady state” and perhaps even more so during acute pain episodes.68, 70 The reduced levels of these regulatory proteins may be a consequence of (a) chronic consumption due to increased thrombin generation resulting from intravascular TF expression and RBC prothrombinase activity; (b) increased binding of protein S by sickle RBCs due to red cell membrane PS exposure71; and/or (c) inhibition of the binding of protein S to β2-glycoprotein 1 by antiphospholipid antibodies,72 resulting in inactivation of protein S by circulating C4b-binding protein. Significantly decreased levels of proteins C and S have been reported in SCD patients who developed thrombotic strokes compared to neurologically normal SCD children.73 Plasma levels of the serine protease inhibitor (SERPIN), heparin cofactor II, are also reported to be decreased in SCD patients.74
Antiphospholipid antibodies are frequently detected in SCD patients.68 In addition, levels of certain subtypes of antiphospholipid autoantibodies, particularly those directed against PS, are markedly elevated in patients with sickle cell anemia, whereas they are normal in patients with HbSC disease.68 In sickle cell anemia patients, a strong correlation exists between antibodies against PS and plasma D-dimers, suggesting a significant role for anti-PS antibodies in coagulation activation in SCD.68
▪ PLATELETS, ENDOTHELIAL CELLS AND MICROPARTICLES
There is ample evidence suggesting that circulating platelets are chronically activated in patients with SCD, a phenomenon that may contribute to the observed coagulation activation in SCD.44 Although there are conflicting reports of platelet survival,75, 76 platelet aggregation responses do appear to be increased in adult patients in the non-crisis, “steady state.”77 This may be explained by an increased number of circulating young, metabolically active platelets, or increased plasma levels of platelet agonists such as thrombin, epinephrine, and adenosine diphosphate. There is increased expression of both CD62P (P-selectin) and CD40L in platelets of SCD patients.67, 78 In addition to elevated plasma levels of the α-granule constituents, TSP, platelet factor 4, and β-thromboglobulin,67 platelet-derived plasma soluble CD40 ligand is elevated in the non-crisis, “steady state,” compared to normal controls.44 The pathogenesis of platelet activation in SCD appears to be due, at least in part, to hemolysis with subsequent decreased bioavailability of NO.79
Circulating platelet aggregates (including both platelet-erythrocyte and platelet-monocyte aggregates) are increased in SCD patients during the non-crisis, “steady state”77, 80, 81 and appear to increase further during acute pain episodes.82 Platelets have also been detected in the heterotypic interaction between the monocyte and reticulocyte in a P-selectin/P-selectin glycoprotein ligand-1-dependent interaction.83 Decreases in platelet lifespan and platelet counts have been reported during acute pain episodes.75, 76 In addition, acute pain episodes appear to be associated with increased platelet activation compared to the non-crisis, “steady state”.44, 68 These findings suggest that decreased platelet survival and increased consumption occur during acute pain episodes.
Increased numbers of circulating endothelial cells have been reported in SCD patients in the non-crisis, “steady state,” with a further increase during acute pain episodes.42 In addition to expressing adhesion markers, these circulating cells abnormally express TF antigen in the “steady state”, and to a greater extent during acute pain episodes.64 Furthermore, these endothelial cells are positive for TF mRNA, with excellent correlation between TF antigen and mRNA expression.64 Circulating MPs, small membrane-derived vesicles released by cells following activation or apoptosis, may be derived from RBCs, platelets, endothelial cells, and monocytes.41 Both the total number of MPs as well as the number of TF-positive MPs are increased in SCD patients, with further increases during acute pain episodes. These TF-positive MPs are derived from endothelial cells and monocytes, but not from RBCs and platelets.84 Plasma markers of coagulation activation, such as D-dimer, TAT, and F1.2, appear to correlate with total MPs, total TF-positive MPs, monocyte-derived TF-positive MPs, and with RBC-derived MPs,84 suggesting a role for MPs in the hypercoagulable state observed in patients with SCD. In a more recent study, the majority of MPs in SCD patients were reported to originate from platelets and erythrocytes, and the numbers of these MPs did not differ significantly between crisis and steady state.85 No MPs originating from monocytes or endothelial cells were detectable and no MPs expressing TF were identified.85 However, erythrocyte-derived MPs correlated strongly with plasma levels of hemolytic markers, as well as with vWF, D-dimer, and F1.2 levels.
▪ THERAPEUTIC IMPLICATIONS
Despite the abundant laboratory evidence of both platelet and coagulation activation in SCD patients, the vast majority of studies using anticoagulants and antiplatelet agents have not provided any convincing benefit in the prevention or treatment of vaso-occlusive complications. As most of these studies have been small and/or poorly controlled, it still remains uncertain whether the observed platelet and coagulation activation observed in these patients contribute to the vascular occlusive episodes that characterize SCD.
Multiple studies of anticoagulants86, 87, 88, 89, 90 (Table 122.1) and antiplatelet agents91, 92, 93, 94, 95, 96 (Table 122.2) have been conducted in SCD patients with varying results. Unfractionated heparin decreases the adhesion of sickle RBCs to endothelium under static conditions, as well as P-selectin-mediated flow adherence of sickle cells to thrombin-treated human vascular endothelial cells.97 A randomized, double-blind, placebo-controlled study of SCD patients during acute pain episodes showed that treatment with the low molecular weight heparin, tinzaparin, resulted in a significant reduction in the overall duration of painful crisis, number of days with the most severe pain scores, and duration of hospitalization compared with placebo.90 The glycoprotein IIb/IIIa inhibitor, eptifibatide, was reported to be safe in a pilot study of sickle cell anemia patients.96 By inhibiting platelet aggregation and decreasing the levels of the inflammatory mediator, soluble CD40 ligand, eptifibatide may be beneficial in the treatment of certain SCD-related complications.96 Table 122.3 shows the rationale for the use of anticoagulants or antiplatelet agents in selected SCD-related complications.
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Blood Coagulation Factors and Inhibitors: Their Primary Structure, Complementary DNAs, Genes, and Expression
Fibrinogen Structure and Function
Megakaryocyte Structure and Platelet Biogenesis
Integrín αIIbβ3 and Platelet Aggregation
Nitric Oxide: The Vascular Biology of Nitric Oxide and Nitric Oxide Synthases
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Blood Coagulation Factors and Inhibitors: Their Primary Structure, Complementary DNAs, Genes, and Expression
Fibrinogen Structure and Function
Megakaryocyte Structure and Platelet Biogenesis
Integrín αIIbβ3 and Platelet Aggregation
Nitric Oxide: The Vascular Biology of Nitric Oxide and Nitric Oxide Synthases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


