Thrombotic and Hemorrhagic Complications Associated With Stem Cell Transplantation
Thrombotic and Hemorrhagic Complications Associated With Stem Cell Transplantation
Yona Nadir
Benjamin Brenner
Thrombotic and hemorrhagic complications are common in stem cell transplantation (SCT) recipients. Endothelial cell (EC) injury is a dominant contributing factor to the hemostatic impairments related to administration of chemotherapy1 and growth factors,1 use of intravenous catheters, and graft-versus-host reaction.2 As bone marrow endothelial progenitor cells are one of the sources for endothelial regeneration, the ability to repair the endothelial damage during SCT is limited. Profound thrombocytopenia is another major contributor to bleeding manifestations. Refractoriness or severe reactions to platelet transfusions, common in patients previously exposed to blood product transfusions during chemotherapy, predispose them to life-threatening bleeding events. A number of alterations in procoagulant, anticoagulant, and, to a lesser extent, fibrinolytic factors have been described. They include prohemorrhagic reduction of factor VII and factor X, as well as tissue-type plasminogen activator (t-PA) increase, simultaneous to prothrombotic decrease of protein C, antithrombin, and plasminogen and increase of fibrinogen, von Willebrand factor (vWF), and plasminogen activator inhibitor (PAI-1).3, 4, 5, 6, 7, 8, 9 Major changes in the coagulation system have been established to be predisposing risk factors for hemostatic complications in SCT patients.
In a retrospective analysis of 364 allogeneic and 83 autologous transplants, 83% of patients presented with at least one hemostatic complication as early as the start of conditioning therapy and throughout a 4- to 10-year follow-up.10 Thromboembolic events, hepatic venoocclusive disease (VOD), and thrombotic microangiopathy (TMA) were most frequently observed in allogeneic transplant recipients, with an incidence of 15%, 4.7%, and 14.6%, respectively, leading to an increased overall mortality.10 The majority of bleeding episodes occurred within the first 4 weeks after transplantation and were relatively mild. However, 27% of the patients hemorrhaged severely, mostly in the gastrointestinal tract and intracranial, doubling the overall mortality of SCT recipients. Bleeding was strongly associated with prolonged thrombocytopenia and graft-versus-host disease (GVHD).10
This chapter deals primarily with thrombotic and hemorrhagic conditions that are specific for SCT. Bleeding resulting from thrombocytopenia and venous thrombosis (e.g., deep vein thrombosis, pulmonary emboli) occurs in SCT patients as well as in many other medical conditions where patients are hospitalized and are not specific. The chapter is organized into two sections (thrombotic and hemorrhagic), each of which considers two special complications. Etiology and treatment of the complications are summarized in Table 103.1.
THROMBOTIC COMPLICATIONS
Site-Specific Microcirculation Thrombosis
The two most common thrombotic manifestations related to SCT, VOD, and TMA are characterized by microcirculation thrombosis. The endothelium synthesizes both anticoagulants and procoagulants. On the anticoagulant side, ECs express tissue factor pathway inhibitor (TFPI), heparan sulfate, thrombomodulin (TM), endothelial protein C receptor (EPCR), t-PA,11, 12, 13 ecto-ADPase, prostacyclin, and nitric oxide (NO). On the procoagulant side, ECs synthesize PAI-1, vWF, proteaseactivated receptors, and tissue factor. Importantly, endothelial derived anticoagulant and procoagulant molecules are not expressed uniformly in the vasculature. For example, TFPI is predominantly expressed in the capillary endothelium, EPCR in large veins and arteries, endothelial NO synthase on the arterial side of the circulation, vWF prevails in veins, and TM in blood vessels of every caliber in all organs apart from the brain.14, 15, 16 These data suggest that ECs originating from different sites of the vascular tree employ site-specific formulas of procoagulants and anticoagulants to balance local hemostasis.11
Hepatic Venoocclusive Disease
VOD is the most common and important regimen-related toxicity experienced after allogeneic and autologous SCT and is characterized by painful hepatomegaly, jaundice, ascites, fluid retention, and weight gain,17, 18, 19, 20 with onset typically prior to day 35 after stem cell reinfusion.17, 18, 19, 20 VOD develops in 1% to 22% of patients after SCT and ranges in severity from a mild, reversible disease to a severe syndrome associated with multiorgan failure and death.18 The incidence and severity of VOD can be influenced by differences in patient characteristics, conditioning regimens, and source of graft, with allogeneic transplantation conferring a threefold increase in risk compared to autologous transplantation.21, 22
Pathogenesis
Hepatic sinusoidal ECs, which comprise 50% of hepatic nonparenchymal cells, are discontinuous. They possess large (100 to 200 nm) membrane-bound, nondiaphragmed cytoplasmic holes or fenestrae, occupying 6% to 8% of the endothelial surface,23 and also display gaps and lack an organized basement membrane. The lack of basement membrane generates a space between sinusoidal ECs and hepatocytes called the space of Disse. The sinusoidal endothelium functions as a selective sieve. The fenestrae act as a dynamic filter for fluids, solutes, and particles, allowing passage of small particles (up to medium-sized chylomicrons) from blood to hepatocytes via the space of Disse. Although the fenestrae are not large enough to accommodate leukocyte transmigration, they do allow cytoplasmic extensions from such cells to penetrate and “touch” the underlying matrix, stellate cells, and hepatocytes. Blood flow velocity in the sinusoids has been estimated to be 400 to 450 µm/s, compared with 500 to 1,000 µm/s in “true capillaries,” thus prolonging interactions between blood and sinusoidal ECs and promoting filtration.23
Table 103.1 Etiology and treatment of thrombotic and hemorrhagic complications in SCT
VOD
TMA
DAH
Hemorrhagic Cystitis
Etiology
Chemotherapy (alkylating agents)/irradiation in SCT21, 22
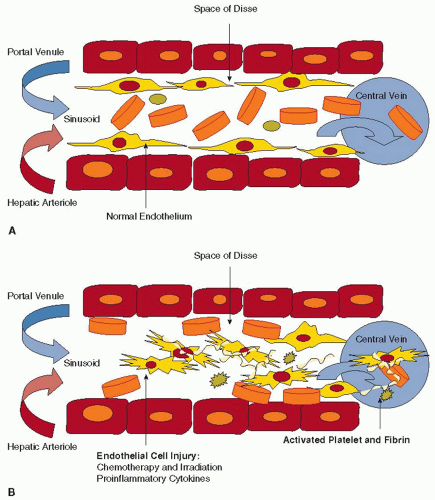
Sinusoidal obstructive syndrome, also known as hepatic VOD, occurs most commonly in response to conditioning regimens (chemotherapy with or without irradiation) used in bone marrow transplantation. The first lesions appear in the sinusoids. Experimental models suggest that exposure to toxins results in swelling and rounding up of sinusoidal ECs, especially in the centrilobular area that has the poorest oxygenation, and is relatively more sensitive to ischemic injury, after which red blood cells enter the space of Disse.24 Here, blood flow essentially dissects the sinusoidal endothelial lining away from the underlying parenchymal cells, resulting in embolization of sinusoidal ECs and occlusion of the downstream venous vessel (FIGURE 103.1A–B, C).24 Subsequent changes include deposition of fibrin within sinusoids and veins, fibroblast cell proliferation, and collagen accumulation in the extracellular matrix (FIGURE 103.1D).25, 26, 27 As the process of venous microthrombosis, ischemia, and fibrosis advances, widespread zonal disruption leads to portal hypertension, hepatorenal syndrome, multiorgan failure, and death.28
Injury to sinusoidal ECs caused by high-dose alkylating agents appears to be the primary event in the pathogenesis of VOD.29, 30, 31 Detoxification of these chemotherapy agents in the liver is mainly mediated by cytochrome P450 and glutathione-S-transferase enzymes, which are present at high concentrations within the centrilobular area.32 Depletion of glutathione stores predisposes hepatocytes to necrosis, whereas addition of glutathione mono-8-diester protects hepatocytes from high-dose alkylator injury.33, 34
Several endothelial injury markers and adhesion molecules are upregulated in patients with VOD, including plasma TM, P- and E-selectins, TFPI, soluble tissue factor, and PAI-1.9, 35In vitro studies show that endothelial PAI-1 production is triggered by cytokine transforming growth factor beta (TGF-β) released from activated platelets.36 Elevated pretransplant plasma levels of TGF-β have been associated with the development of hepatic VOD.37 Additionally, proinflammatory cytokines could also contribute to the initial endothelial injury, and increased tumor necrosis factor (TNF-α), interleukin (IL)-6, IL-8, and IL-1β are correlated with the jaundice, renal dysfunction, and pulmonary disease of SCT.4, 38
The role of thrombophilic factors in the pathogenesis of thrombotic complications in SCT is unclear. In a small retrospective study, the prothrombin gene 20210 G-A mutation was found to be a predisposing factor for VOD,39 and a prospective study in children revealed a strong association between factor V Leiden mutation and VOD.40 More studies are warranted to confirm the association between thrombophilia and VOD.
Therapy
The most established practice in VOD prevention has been the use of pharmacokinetics to monitor chemotherapeutic drug levels to minimize hepatic injury. Prophylactic administration of ursodeoxycholic acid, a hydrophilic water-soluble bile acid, has been studied in randomized placebo-controlled trials, some showing a statistically significant benefit in patients predicted to be at a high risk of VOD.41, 42, 43 However, a large phase III study failed to demonstrate significant benefit of this approach.44 The role of heparin anticoagulation, alone or in combination with other agents, in VOD prophylaxis has been assessed in only one randomized study, which reported a beneficial effect of low-dose continuous heparin infusion.45 Low molecular weight heparin is relatively safe and may have some impact on VOD prevention,46, 47, 48, 49 but well-designed studies are needed to confirm these results.
There is no approved therapy for established hepatic VOD. Current approaches focus on supportive care and on anticoagulant or fibrinolytic drugs,45, 46, 50, 51, 52 based on pathologic findings of hepatic venules or sinusoids obliteration by fibrin.18, 26, 27 The major disadvantage of exposure to heparin or recombinant t-PA is the high risk of life-threatening bleeding in patients with concurrent thrombocytopenia. The lack of response to platelet transfusions due to portal hypertension, splenic sequestration, and antiplatelet antibodies hampers effective management.51, 53
FIGURE 103.1 Mechanism of VOD. A: Normal blood flow in hepatic sinusoid. No basement membrane is present. B: Damaged ECs torn away from the sinusoid. Red blood cells enter the potential space of Disse and aggravate EC separation. Damaged ECs and the exposure of the subendothelial space activate platelets and the coagulation system to form fibrin.
Defibrotide, a single-stranded polydeoxyribonucleotide that exerts antithrombotic activity by binding to vascular endothelium, has shown promising results in VOD management.54, 55, 56 Defibrotide upregulates the endothelial release of prostacyclin (PG I2), prostaglandin E2, TM, and t-PA,57, 58, 59 and decreases thrombin generation, tissue factor expression, PAI-1 release, and endothelin activity.57, 60 Defibrotide exerts no significant effect on systemic coagulation,61 but it has antiangiogenic potential in ECs and in an animal model.62 Preclinical studies have demonstrated profibrinolytic effects and inhibition of fibrin deposition, with selective activity in small vessels.63 Initial clinical reports of defibrotide for severe VOD recorded complete resolution in 36% to 42% of patients, with most individuals surviving beyond day 100.54, 55, 64 In a recent randomized phase II dose-finding trial in adult and pediatric patients, defibrotide 25 mg/kg/d was selected for forthcoming phase III trials.65 Preemptive antithrombin replacement and combined antithrombin/defibrotide therapy allowed excellent remission and survival rates in a prospective case series study of pediatric SCT patients.66 In another report on 58 adults undergoing SCT, no patient developed VOD following the use of defibrotide prophylaxis (without concurrent heparin).67 Cappelli et al.68 reported on 57 children with beta thalassemia at a very high risk for developing VOD (liver fibrosis, iron overload, hepatitis C virus infections, busulfan-based conditioning), and only one patient developed VOD, after discontinuation of defibrotide 6 days prior to VOD onset. To date, no phase III randomized studies have been published on the use of defibrotide as prophylaxis or treatment of VOD.
Only gold members can continue reading. Log In or Register to continue
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Inherited Thrombocytopenias
Inherited Thrombocytopenias
 Unusual Sites of Arterial Occlusion
Unusual Sites of Arterial Occlusion
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient



