Thrombohemorrhagic Complications of Sepsis
Marcel Levi
Sepsis is a clinical syndrome that is caused by an infection, often associated with bacteremia and characterized by the presence of systemic signs and symptoms of inflammation.1 When sepsis leads to organ failure, the term severe sepsis is used. The incidence of sepsis is estimated to be about 2.5 per 1,000 in the Western world and shows a rapid 8.7% annual increase over the last 20 years.2 Total inhospital mortality of sepsis is around 20%, whereas severe sepsis is associated with mortality rates of 40% to 50%.3 Treatment of sepsis is focused on adequate antibiotic therapy, source control, and appropriate supportive care and organ function replacement, if required.
Virtually all patients with sepsis have coagulation abnormalities. These abnormalities range from subtle activation of coagulation that can only be detected by sensitive markers for coagulation factor activation to somewhat stronger coagulation activation that may be detectable by a small decrease in platelet count and prolongation of global clotting times to fulminant disseminated intravascular coagulation (DIC), characterized by simultaneous widespread microvascular thrombosis and profuse bleeding.4, 5 Septic patients with severe DIC may present with manifest thromboembolic disease or clinically less apparent microvascular fibrin deposition and multiple organ dysfunction.6, 7, 8 Alternatively, severe bleeding may be the leading symptom,9 but quite often a patient with DIC has simultaneous thrombosis and bleeding. Bleeding is caused by consumption and subsequent exhaustion of coagulation proteins and platelets, due to the ongoing activation of the coagulation system.10 In its most severe form, this combination may present as the Waterhouse-Friderichsen syndrome as seen during fulminant septicemia.11
INCIDENCE OF COAGULATION ABNORMALITIES IN SEPSIS
Clinically relevant coagulation abnormalities may occur in 50% to 70% of patients with sepsis, whereas about 35% of patients will meet the criteria for DIC.1, 12 In general, the incidence of thrombocytopenia (platelet count <150 × 109/L) in critically ill medical patients is 35% to 50%.13, 14, 15 Typically, the platelet count correlates with sepsis severity, decreasing during the first 4 days of illness.16, 17 The factors that contribute to thrombocytopenia are impaired platelet production and increased consumption, destruction, or sequestration. Although the high levels of platelet production-stimulating proinflammatory cytokines and thrombopoietin circulate,18 marked hemophagocytosis by monocytes and macrophages19 and thrombin-induced platelet activation and destruction at the endothelial cell surface explain the thrombocytopenia.20 A prolonged global coagulation time (prothrombin time [PT] or activated partial thromboplastin time [aPTT]) occurs in 14% to 28% of patients.21, 22 Other coagulation abnormalities include high fibrin split products in virtually all patients with sepsis23, 24, 25 and decreased levels of coagulation inhibitors, such as antithrombin-3 (AT3) and protein C in 90% of sepsis patients.25, 26
RELEVANCE OF COAGULATION ABNORMALITIES IN PATIENTS WITH SEPSIS
There is strong evidence that activation of coagulation in concert with inflammatory activation can result in microvascular thrombosis, which contributes to multiple organ failure in patients with severe sepsis.27 Autopsy findings include diffuse bleeding and microthrombi in small blood vessels and thrombi in midsize and larger arteries and veins, in association with ischemia and necrosis.28, 29, 30 Further, experimental bacteremia or endotoxemia causes intra- and extravascular fibrin deposition in kidneys, lungs, liver, and brain, and interventions in these experimental models improve organ failure and, in some cases, mortality.31, 32, 33, 34, 35 Last, clinical studies support the notion of coagulation as an important denominator of clinical outcome, DIC being an independent predictor of organ failure and mortality in patients with sepsis,6, 36 associated with mortality in 43%, as compared with 27% in patients without DIC.37
Thrombocytopenia in patients with sepsis is related to an increased risk of bleeding, those with a platelet count of <50 × 109/L having a four- to fivefold higher risk than patients with a higher platelet count.13, 15 The overall risk of intracerebral bleeding in ICU patients with sepsis is relatively low (0.3% to 0.5%). However 88% of patients with intracranial bleeding have platelet counts <100 × 109/L.38 Regardless of the cause, thrombocytopenia is an independent predictor of ICU mortality in multivariate analyses, with a relative risk of 1.9 to 4.2.13, 15, 39 In particular, a sustained thrombocytopenia for more than 4 days after ICU admission or a drop in platelet count of >50% during ICU stay is related to a four- to sixfold increase in mortality.13, 16 The platelet count is a stronger predictor for ICU mortality than composite scoring systems, such as the Acute Physiology and Chronic Health Evaluation (APACHE) II score or the Multiple Organ Dysfunction Score. Prolonged global coagulation tests to >1.5 times control also predict excessive bleeding and increased mortality.21, 22
PATHOGENETIC PATHWAYS IN THE COAGULOPATHY OF SEPSIS
Various mechanisms at different sites in the hemostatic balance act simultaneously toward a procoagulant state (FIGURE 123.1), the most important mediators of which are cytokines.40 There
is extensive cross-talk between inflammation and coagulation,41 and interestingly, systemic activation of coagulation and inflammation in sepsis can have some organ-specific manifestations.42
is extensive cross-talk between inflammation and coagulation,41 and interestingly, systemic activation of coagulation and inflammation in sepsis can have some organ-specific manifestations.42
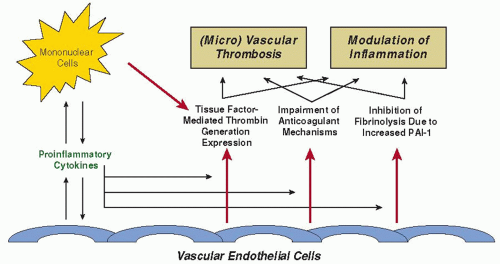 FIGURE 123.1 Schematic representation of pathogenetic pathways involved in the activation of coagulation in sepsis. During sepsis, both perturbed endothelial cells and activated mononuclear cells may produce proinflammatory cytokines that mediate coagulation activation. Activation of coagulation is initiated by TF expression on activated mononuclear cells and endothelial cells. In addition, downregulation of physiologic anticoagulant mechanisms and inhibition of fibrinolysis by endothelial cells will further promote intravascular fibrin deposition. PAI-1; plasminogen activator inhibitor type 1. |
Initiation of Coagulation Activation
Systemic activation of coagulation in patients with sepsis has been considered to be a result of direct activation of the contact system by microorganisms or endotoxin.43 However, it has become apparent that the principal initiator of thrombin generation in sepsis is tissue factor (TF), without significant change in markers reflecting activation of the contact system.44, 45 Furthermore, abrogation of the TF/factor VII(a) pathway by monoclonal antibodies completely inhibits thrombin generation in endotoxin-challenged chimpanzees and prevents DIC and mortality in baboons that are infused with Escherichia coli33, 46, 47
TF is a transmembrane 45 kDa protein, that is constitutively expressed on a number of cells throughout the body.48 The majority of these cells are in tissues not in direct contact with blood, such as the adventitial layer of large blood vessels. However, TF gets into contact with blood upon disruption of the vascular integrity, or if cells present in the circulation are altered to express TF. In sepsis, circulating mononuclear cells, stimulated by proinflammatory cytokines, express TF, but other than in severe meningococcemia,49 it is difficult to demonstrate ex vivo TF expression on monocytes of septic patients or experimental animals. However, low dose endotoxemia in healthy subjects results in a 125-fold increase in monocyte TF mRNA levels.50 Another source of TF may be on polymorphonuclear cells,51 although it is unlikely that these cells actually synthesize TF in substantial quantities.52 Based on the transfer of TF from leukocytes to activated platelets in an ex vivo perfusion system, it is hypothesized that this “blood borne” TF is transferred between cells through microparticles derived from activated mononuclear cells.53
Platelets play a pivotal role in the pathogenesis of coagulation abnormalities in sepsis. Platelets can be activated directly by proinflammatory mediators, such as platelet activating factor.54 Once thrombin forms, additional platelets are activated, which then accelerates further fibrin formation. The expression of P-selectin on the platelet membrane not only mediates the adherence of platelets to leukocytes and endothelial cells but also enhances the expression of TF on monocytes by nuclear factor kappa-B (NFκB) activation.55 P-selectin is shed from the platelet membrane and soluble P-selectin levels are increased during systemic inflammation.55
Impairment of the Antithrombin, Protein C, and TFPI Anticoagulant Pathways in Sepsis
Activation of coagulation is regulated by three major anticoagulant pathways, namely, AT3, the protein C system, and TF pathway inhibitor (TFPI). During sepsis-induced activation of coagulation, the function of all three pathways can be impaired (FIGURE 123.2).
AT3 is a serine protease inhibitor and the main inhibitor of thrombin and factor Xa. During severe inflammatory responses, AT3 levels are markedly decreased due to consumption (as a result of ongoing thrombin generation), impaired synthesis (as a result of a negative acute phase response), and degradation by elastase from activated neutrophils.56, 57 A reduction in
glycosaminoglycan availability at the endothelial surface (due to the influence of proinflammatory cytokines on endothelial synthesis) also contributes to reduced AT3 function, since glycosaminoglycans act as physiologic heparin-like cofactors of AT3. Binding of glycosaminoglycans to AT3 induces a conformational change at the reactive center of the molecule, thereby rendering this protease inhibitor from a slow to a very efficient inhibitor.58 Prospective clinical studies in patients at high risk for sepsis show that a marked decrease in levels of AT3 precedes the clinical manifestation of the infection, which may indicate that AT3 is involved in the early stages of coagulation activation during sepsis.59
glycosaminoglycan availability at the endothelial surface (due to the influence of proinflammatory cytokines on endothelial synthesis) also contributes to reduced AT3 function, since glycosaminoglycans act as physiologic heparin-like cofactors of AT3. Binding of glycosaminoglycans to AT3 induces a conformational change at the reactive center of the molecule, thereby rendering this protease inhibitor from a slow to a very efficient inhibitor.58 Prospective clinical studies in patients at high risk for sepsis show that a marked decrease in levels of AT3 precedes the clinical manifestation of the infection, which may indicate that AT3 is involved in the early stages of coagulation activation during sepsis.59
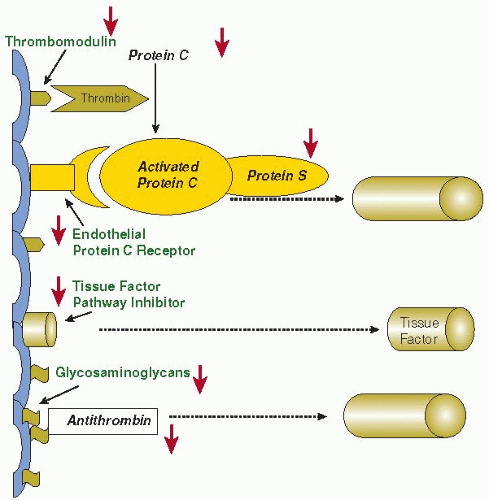 FIGURE 123.2 Schematic representation of the three important physiologic anticoagulant mechanisms and their point of impact in the coagulation system. In sepsis, these mechanisms are impaired by various mechanisms (red arrows). The protein C system is dysfunctional due to low levels of zymogen protein C, downregulation of TM and the EPCR, and low levels of free protein S due to acute phase-induced high levels of its binding protein, that is, C4b-binding protein. There is a relative insufficiency of the endothelial cell-associated TFPI. The antithrombin system is defective due to low levels of antithrombin and impaired glycosaminoglycan expression on perturbed endothelial cells. |
Endothelial dysfunction is even more important in the impairment of the protein C system during inflammation. Under physiologic conditions, protein C is activated by thrombin bound to the endothelial cell membrane-associated thrombomodulin (TM). TM is a membrane protein with several domains, including a lectin-like domain, six epidermal growth factor (EGF)-like repeats, a transmembrane domain and a short cytoplasmatic tail.60 The binding of thrombin to TM occurs at the site of the EGF-repeats.61 This binding not only results in a 100-fold increase in the activation of protein C, but also blocks thrombin-mediated conversion of fibrinogen to fibrin and inhibits thrombin binding to other receptors on platelets and inflammatory cells. TM accelerates the activation of the plasma carboxypeptidase thrombin-activatable fibrinolysis inhibitor (TAFI), an important inhibitor of fibrinolysis.62 Activated protein C (APC) regulates coagulation activation by proteolytic cleavage of the essential cofactors Va and VIIIa. Binding of protein C to the endothelial protein C receptor (EPCR) results in a fivefold augmentation of protein C activation by the TM-thrombin complex.63 However, during sepsis, in addition to low levels of protein C due to impaired synthesis56 and degradation by neutrophil elastase,64 the protein C system is defective due to downregulation of TM at the endothelial surface, mediated by the proinflammatory cytokines tumor necrosis factor-alpha (TNF-α) and IL-1β.65
Observations in patients with severe Gram-negative septicemia confirm the downregulation of TM in vessels with and without thrombosis and impaired activation of protein C in vivo.66 Low levels of free protein S (the cofactor of APC) may further compromise function of the protein C system. In plasma, 60% of protein S is complexed to a complement regulatory protein, C4b binding protein (C4bBP). Increased plasma levels of C4bBP as a consequence of the acute phase reaction in inflammatory diseases may result in a relative free protein S deficiency. Although the β-chain of C4bBP (which mainly governs the binding to protein S) is not much affected during the acute phase response,67 C4bBP infusion increases organ dysfunction and mortality in septic baboons.68 Animal experiments of severe inflammation-induced coagulation activation convincingly show that compromising the protein C system results in increased morbidity and mortality, whereas restoring an adequate function of APC improves survival and organ failure.69 Interestingly, experiments in mice with a one-allele targeted deletion of the protein C gene (resulting in heterozygous protein C deficiency) have more severe DIC and organ dysfunction and a higher mortality than wild type littermates.70
A third inhibitory mechanism of thrombin generation involves TFPI, the main inhibitor of the TF-factor VIIa complex. TFPI is a complex multidomain Kunitz-type protease
inhibitor that binds to the TF-factor VIIa complex and factor Xa.71 The TFPI-factor Xa complex binds to negatively charged membrane surfaces, which may increase the local concentration of TFPI at cellular sites and facilitate inhibition of membranebound TF-factor VIIa complex. The role of TFPI in the regulation of inflammation-induced coagulation activation is not completely clear. Administration of recombinant TFPI (r-TFPI) (and thereby achieving higher than physiologic plasma concentrations of TFPI) blocks inflammation-induced thrombin generation in humans, and pharmacologic doses of TFPI prevent mortality during systemic infection and inflammation.31, 72 However, the endogenous concentration of TFPI is presumably insufficient to regulate coagulation activation and downstream consequences during systemic inflammation, as confirmed in patients with sepsis.73, 74
inhibitor that binds to the TF-factor VIIa complex and factor Xa.71 The TFPI-factor Xa complex binds to negatively charged membrane surfaces, which may increase the local concentration of TFPI at cellular sites and facilitate inhibition of membranebound TF-factor VIIa complex. The role of TFPI in the regulation of inflammation-induced coagulation activation is not completely clear. Administration of recombinant TFPI (r-TFPI) (and thereby achieving higher than physiologic plasma concentrations of TFPI) blocks inflammation-induced thrombin generation in humans, and pharmacologic doses of TFPI prevent mortality during systemic infection and inflammation.31, 72 However, the endogenous concentration of TFPI is presumably insufficient to regulate coagulation activation and downstream consequences during systemic inflammation, as confirmed in patients with sepsis.73, 74
Inhibition of Endogenous Fibrinolysis in Sepsis
Experimental models indicate that at the time of maximal activation of coagulation in sepsis, the fibrinolytic system is largely shut off. The acute fibrinolytic response to inflammation is the release of plasminogen activators, in particular tissue-type plasminogen activator and urokinase-type plasminogen activator, from storage sites in vascular endothelial cells. However, this increase in plasminogen activation and subsequent plasmin generation is counteracted by a delayed but sustained increase in plasminogen activator inhibitor type 1 (PAI-1).75, 76 The resulting effect on fibrinolysis is complete inhibition and, as a consequence, inadequate fibrin removal, thereby contributing to microvascular thrombosis. Experiments in mice with targeted disruptions of genes encoding components of the plasminogen-plasmin system confirm that fibrinolysis plays a major role in inflammation-induced coagulation. Mice with a deficiency of plasminogen activators have more extensive fibrin deposition in organs when challenged with endotoxin, whereas PAI-1 knockout mice, in contrast to wild-type controls, have no microvascular thrombosis upon endotoxin.77, 78 A functional mutation in the PAI-1 gene, the 4G/5G polymorphism, not only influences the plasma levels of PAI-1, but also is linked to significantly higher plasma PAI-1 concentrations and an increased risk of death.79 The PAI-1 polymorphism does not influence the risk of contracting meningitis, but probably increases the likelihood of developing septic shock from meningococcal infection.80
Regulatory Role of Cytokines in the Coagulopathy of Sepsis
Most proinflammatory cytokines activate coagulation in vitro. In patients with sepsis high levels of cytokines are detectable in the circulation, and experimental bacteremia or endotoxemia results in the transient enhancement of serum cytokine levels.81 TNF is detectable first, followed by an increase in levels of interleukin-6 (IL-6) and IL-1 (Table 123.1). The hypothesis that TNF plays an important role in coagulation activation in vivo is strengthened by studies in which cancer patients or healthy human volunteers injected with purified recombinant TNF showed coagulation activation that is virtually identical to endotoxin-induced effects on coagulation.45, 82 However, blocking TNF activity completely abolished endotoxin-induced increase in TNF but coagulation activation was unaffected.83, 84 Also, in baboons infused with a lethal dose of E. coli, treatment with an anti-TNF antibody had little or no effect on fibrinogen consumption.85 Moreover, clinical studies in septic patients with an anti-TNF monoclonal antibody did not show a beneficial effect of this treatment.86
Table 123.1 Most important cytokines governing the interaction between inflammation and coagulation | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
These observations forced a reconsideration of the role of TNF as principal mediator of endotoxin-induced activation of coagulation. Subsequent studies of IL-6 showed that infusion of a monoclonal anti-IL-6 antibody resulted in the complete abrogation of endotoxin-induced activation of coagulation in chimpanzees.87 In addition, studies in cancer patients receiving recombinant IL-6 indicated that thrombin is generated following the injection of this cytokine.88 These data suggest that IL-6 rather than TNF is the relevant mediator for the procoagulant response in DIC.
While IL-1 is a potent agonist of TF expression in vitro, its role has not been clarified in vivo. Administration of an IL-1 receptor antagonist partly blocks the procoagulant response in a baboon sepsis model and treatment of patients with an IL-1 receptor inhibitor reduces thrombin generation.89, 90, 91 However, most of the procoagulant changes after an endotoxin challenge occur well before IL-1 becomes detectable in the circulation, leaving a potential direct role of IL-1 in coagulation activation in sepsis an unresolved issue. Anti-inflammatory cytokines, such as IL-10, may modulate coagulation activation. Infusion of recombinant human IL-10 completely blocks the endotoxin-induced changes in coagulation and fibrinolysis in human volunteers,92 but its relevance in the pathogenesis of the sepsisassociated coagulopathy remains to be established.
Cross-Talk Between Coagulation and Inflammation in Sepsis
Coagulation proteases and protease inhibitors not only interact with coagulation protein zymogens, but also with specific cell receptors to induce signaling pathways. In particular, protease interactions that affect inflammatory processes may be important in sepsis. Coagulation of whole blood in vitro results in a detectable expression of IL-1β mRNA in blood cells,93 and thrombin markedly enhances endotoxin-induced IL-1 activity in culture supernatants of guinea pig macrophages.94 Similarly, clotting blood produces IL-8 in vitro.95 Factor Xa, thrombin, and fibrin can also activate endothelial cells, eliciting the synthesis of IL-6 and/or IL-8.96, 97 The most important mechanisms by which coagulation proteases influence inflammation is by binding to the so-called protease-activated receptors (PARs), of which four types (PAR 1 to 4) have been identified, all belonging to the family of transmembrane domain, G-protein-coupled receptors.98 A peculiar feature of PARs (in contrast to most other receptors of the superfamily) is that they serve as their own ligand. Proteolytic cleavage by an activated coagulation factor leads to exposure of a neoamino-terminus that activates the same receptor, initiating transmembrane signaling. PARs 1, 3, and 4 are thrombin receptors whereas PAR-2 (and PAR-1) can be activated by the TF-factor VIIa complex and factor Xa. PARs are localized in the vasculature on endothelial cells, mononuclear cells, platelets, fibroblasts, and smooth muscle cells.98 Binding of thrombin and TF-factor VIIa to its receptor in macrophages affects neutrophil infiltration and proinflammatory cytokine (TNF-α, IL-1β) expression.99, 100 Administration of recombinant factor VIIa to healthy human subjects causes a three- to fourfold rise in plasma levels of IL-6 and IL-8.101
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Inherited Thrombocytopenias
Inherited Thrombocytopenias
 Unusual Sites of Arterial Occlusion
Unusual Sites of Arterial Occlusion
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


