The Thrombotic Thrombocytopenic Purpura and Hemolytic Uremic Syndromes
The Thrombotic Thrombocytopenic Purpura and Hemolytic Uremic Syndromes
James N. George
Thrombotic thrombocytopenic purpura (TTP)1,2 and the hemolytic uremic syndrome (HUS)3,4 were described initially as distinct disorders. Although they may be distinct in their classic presentations, TTP in adults with acquired, severe ADAMTS13 deficiency and the diarrhea-associated typical HUS in children, in many patients this distinction is neither apparent nor important. TTP is often said to be associated with more severe neurologic abnormalities and HUS with more severe renal failure. However, many adult patients with acquired, severe ADAMTS13 deficiency have neither severe neurologic abnormalities nor renal failure,5,6 and some children with typical HUS whose primary abnormality is acute renal failure have severe neurologic abnormalities.7,8,9 There are multiple other syndromes that are described as TTP or HUS in addition to these two classic disorders in which the distinction between TTP and HUS is similarly unclear. Table 99.1 summarizes the common clinical nomenclature of these syndromes.6
Pathologic abnormalities are similar in both TTP and HUS. The characteristic histologic features of thrombi in terminal arterioles and capillaries are described as thrombotic microangiopathy (TMA) (Table 99.1).10,11FIGURE 99.1 demonstrates the characteristic pathologic features of systemic TMA in a patient with acquired, severe ADAMTS13 deficiency. TMA can also occur in multiple other disorders, such as malignant hypertension, scleroderma, antiphospholipid antibody syndrome, acute renal allograft rejection, and severe preeclampsia and the hemolysis, elevated liver enzymes, and a low platelet count (HELLP) syndrome10; in these disorders the TMA is usually restricted to the kidney. Microangiopathic hemolysis with red-cell fragmentation may result from the turbulence and high shear stress of circulation through partially occluded arterioles.12 Therefore, the clinical features of all disorders associated with TMA are similar. All disorders associated with TMA may resemble the clinical features of TTP and HUS. This can make clinical decisions of definition, diagnosis, and treatment of the TTP and HUS syndromes difficult.
The definition of TTP has changed since the classic review in 1966 that summarized all previously reported cases.13 This review defined a pentad of clinical features that became the cornerstone of diagnosis of TTP for many years: (a) microangiopathic hemolytic anemia, (b) thrombocytopenia, (c) neurologic symptoms and signs, (d) renal function abnormalities, and (e) fever. In that review,13 the diagnosis of TTP was supported by pathologic demonstration of hyaline thrombi in 93% of patients; 90% of the patients died. In the past 30 years, the availability of curative plasma exchange treatment14,15 has created an urgency for establishing the diagnosis of TTP, which in turn has resulted in less stringent diagnostic criteria.6,15 Now only microangiopathic hemolytic anemia and thrombocytopenia without another clinically apparent cause are sufficient to establish the diagnosis of TTP.6,15 Decreased stringency of diagnostic criteria has inevitably led to an increased frequency of diagnosis16 and to a broader clinical spectrum of disorders for which plasma exchange treatment is considered. A current classification of presenting clinical features and disease associations of the TTP and HUS syndromes is presented in Table 99.2.
This chapter focuses on TTP in adults, initially describing adults with acquired, severe ADAMTS13 deficiency (defined as <10% activity) (Table 99.2). Clinical characteristics of the TTP and HUS syndromes that are associated with other apparent etiologies are discussed in subsequent sections.
TTP ASSOCIATED WITH ACQUIRED, SEVERE ADAMTS DEFICIENCY
Although patients with acquired, severe ADAMTS13 deficiency (activity <10%) are considered to be the “classic” TTP, they have represented only 23% of all 299 patients who were treated with plasma exchange for an initial clinical diagnosis of TTP or HUS in the Oklahoma TTP-HUS Registry, 1995 to 2010, and who had measurement of ADAMTS13 activity immediately before plasma exchange was begun (FIGURE 99.2).5,6 Only 48% of 114 patients with idiopathic TTP and severe ADAMTS13 deficiency.6 This observation emphasizes the heterogeneity of the TTP and HUS syndromes. Although patients with acquired, severe ADAMTS13 deficiency constitute only a minority of all patients with these syndromes, their pathogenesis, clinical features, and outcomes are well described and represent the basis for understanding the mechanisms of microvascular thrombosis as well as the evaluation and management of patients with clinically suspected TTP.
Pathogenesis
von Willebrand factor (vWF) abnormalities are central to the pathogenesis of TTP in patients with acquired, severe ADAMTS13 deficiency.17,18,19,20,21,22 vWF is secreted by endothelial cells into the plasma where it is processed into smaller multimers that do not spontaneously interact with circulating platelets (see Chapter 13). ADAMTS13, an enzyme present in normal plasma, processes vWF by proteolysis, which is the principal mechanism in this respect. vWF may also be processed following secretion by reduction of disulfide bonds that link the vWF dimers.23,24,25 In 1982, Moake et al. observed in the plasma of patients with chronic relapsing (congenital) TTP26 unusually large vWF multimers (ULvWF), which were comparable in size to vWF initially secreted by endothelial cells. These ULvWF multimers can directly agglutinate platelets when the vWF structure is altered by the high shear stress that occurs in arterioles.20 In 1996 to 1998, the origin of ULvWF was documented to be a severe deficiency of ADAMTS13 activity.17,18,27,28,29,30 In patients with acquired TTP, autoantibodies inhibit ADAMTS13 function by interacting with multiple domains, particularly the Cysrich and spacer domains.31 In some patients with apparently acquired, severe ADAMTS13 deficiency, no inhibitory autoantibodies are detectable. Autoantibodies may have been neutralized by previous transfusions,5 or autoantibodies may impair ADAMTS13 function by accelerating clearance from plasma or by impairing binding to the endothelial cell surface rather than by neutralizing enzymatic activity.32
Table 99.1 Nomenclature of the TTP and HUS syndromes
Name
Definition
Pathologic Descriptive Term
TMA
The characteristic histologic abnormality of both TTP and HUS. Describes the microvascular features (swelling of endothelial cells and the subendothelial space) associated with thrombosis. TMA may also occur in many other disorders, such as malignant hypertension, antiphospholipid antibody syndrome, systemic lupus erythematosus, and preeclampsia.
Acquired Clinical Syndromes
TTP
The appropriate term for all adults with microangiopathic hemolytic anemia and thrombocytopenia with or without renal failure or neurologic abnormalities and without an alternative etiology. Children without renal failure are also described as TTP. Includes but is not limited to patients with severe ADAMTS13 deficiency (<10%). In some adults with predominant acute renal failure, such as with quinine sensitivity or E. coli O157:H7 infection, the comprehensive term, TTP-HUS, is often used.
Typical HUS
Children with a syndrome of microangiopathic hemolytic anemia, thrombocytopenia, and renal failure with a diarrhea prodrome caused by enteric infection with E. coli O157:H7 or other Shiga toxin-producing bacteria.
aHUS
A syndrome of microangiopathic hemolytic anemia, thrombocytopenia, and renal failure without a diarrhea prodrome. May be caused by pneumococcal infections or acquired disorders of complement regulation.
Congenital Clinical Syndromes
Upshaw-Shulman syndrome
Caused by congenital deficiency of ADAMTS13.
aHUS
Associated with congenital deficiencies of complement regulatory factors, resulting in unrestrained complement activation.
Adapted from George JN. How I treat patients with thrombotic thrombocytopenic purpura. Blood 2010;116:4060-4069.
Undetectable ADAMTS13 activity is not always associated with evidence for disease. Approximately 20% of patients have persistent severe deficiency of ADAMTS13 activity following recovery from an acute episode of TTP.5 These observations are similar to those of the absence of the TTP phenotype in Adamts13-/- mice33,34 and suggest that absence of ADAMTS13 may be only one of several components causing acute episodes of TTP. However in baboons, it has been demonstrated that absence of ADAMTS13 activity is sufficient to initiate the clinical features of TTP.22
Risk Factors for TTP Associated with Acquired, Severe ADAMTS13 Deficiency
Similar to other thrombotic and autoimmune disorders, multiple risk factors may contribute to the onset of TTP. Similar to systemic lupus erythematosus (SLE),35,36 middle age, female gender, and black race are important risk factors.36,37,38 Although it had been postulated that blood group O may be protective against TTP because it is associated with lower plasma vWF levels39,40,41,42,43 and a faster rate of vWF proteolysis by ADAMTS13,42,44 blood group O is actually a risk factor for TTP associated with acquired, severe ADAMTS13 deficiency.45 This observation suggests a novel hypothesis that higher basal levels of normal-sized vWF in people with non-O blood groups may compete with ULvWF for platelet binding, reducing the risk for ULvWF-mediated formation of platelet thrombi and providing partial protection from ULvWF-mediated thrombosis in patients with non-O blood groups. This hypothesis is consistent with previous observations that elevated plasma vWF levels are not a risk factor for the occurrence of the features of TTP in Adamts13-/- mice.33
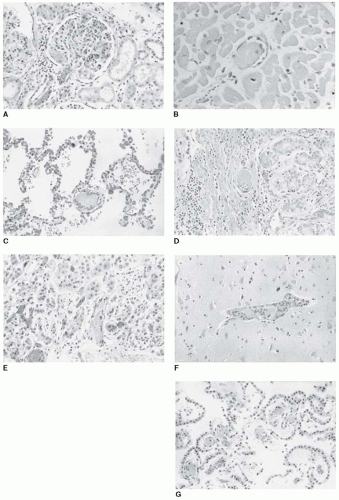
FIGURE 99.1 Systemic microvascular thrombi in TTP. These autopsy tissue sections are from a previously healthy young man who became ill with 3 days of abdominal pain, nausea, and vomiting followed by severe confusion. Clinical features on admission to the hospital were platelet count 11 × 109/L, hematocrit 21%, LDH 2,231 U/L, creatinine 1 mg/dL, urinalysis with proteinuria and hematuria, and no fever. He received three plasma exchange treatments plus corticosteroids but died with grand mal seizures and cardiopulmonary arrest 3 days after hospital admission. Subsequent analysis of serum collected before the initial plasma exchange treatment demonstrated ADAMTS13 activity <5% with an ADAMTS13 inhibitor of >2 Bethesda units. Tissue sections demonstrate (A) kidney afferent arteriole and glomerular capillary thrombi, (B) myocardial thrombi, (C) pulmonary thrombi, (D) gastric submucosal thrombi, (E) adrenal thrombi, (F) cerebral thrombi, and (G) thrombi in the choroid plexus. (Photographs by Zoltan Laszik, MD, Department of Pathology, University of Oklahoma.)
Table 99.2 Clinical features of the TTP and HUS syndromes
Syndrome
Clinical Features
Acquired Disorders
ADAMTS13 activity <10%
Considered to be the “classic” TTP. Autoimmune ADAMTS13 deficiency occurs primarily in adult women with increased relative frequency among blacks. Acute episodes may be triggered by pregnancy or infections and may be associated with SLE or other autoimmune disorders. One-third has no neurologic abnormalities; acute renal failure and fever rare; immunosuppressive treatment required in addition to PEX. Relapse may occur in 41%.
Idiopathic TTP, ADAMTS13 activity ≥10%
Etiology unknown. Some may be associated with abnormalities of complement regulation. Compared to patients with ADAMTS13 activity <10%, these patients are older, more often have renal failure, and do not relapse. PEX appropriate because clinical distinction from patients with ADAMTS13 <10% is uncertain.
Typical HUS
Primarily occurs in children <5 years old and accounts for 90%-95% of childhood HUS; also occurs in adults. Preceded by diarrhea, often bloody, and manifested by acute renal failure. Severe neurologic abnormalities uncommon in children but more common in adults. Children managed by supportive care; PEX appropriate for adults because clinical distinction from patients with ADAMTS13 <10% is uncertain. Relapse does not occur.
aHUS
Patients in whom HUS is not preceded by diarrhea. Accounts for 5%-10% of childhood HUS; also occurs in adults. PEX may be appropriate. Relapse may occur.
Drug-induced: acute, immune mediated
Quinine is the most common and the only one with clearly documented etiology, caused by drug-dependent antibodies to platelets, endothelial cells, and other cells. Characterized by a sudden onset of systemic symptoms and anuric acute renal failure. PEX appropriate because drug etiology may be uncertain. Also reported with other drugs.a
Drug-induced: dose-dependent toxicity
Mitomycin C, gemcitabine, and cyclosporine are the most commonly reported drugs. Also associated with drugs inhibiting VEGF. Insidious onset with predominant renal failure. Reversibility and role of PEX are uncertain.
Congenital Disorders
Upshaw-Schulman syndrome (ADAMTS13 deficiency)
Initial episodes may occur at any age; recurrent acute episodes are common. Plasma infusion is sufficient treatment; prophylactic plasma infusion may be required.
Deficiencies of complement regulatory factors (aHUS)
Described as aHUS since acute renal failure is predominant without a diarrhea prodrome. Initial acute episodes may occur at any age. PEX may be appropriate. Relapse may occur.
a Other drugs that have been associated with acute and apparently immune-mediated TTP and HUS syndromes are clopidogrel, ticlopidine, and trimethoprim-sulfamethoxazole.
In addition to demographic risk factors, there are multiple acquired risk factors that can be associated with or perhaps even trigger acute episodes of TTP in susceptible patients. Pregnancy is associated with episodes of both congenital and acquired TTP.46,47,48 The increased risk for TTP during pregnancy may be related to the decreased ADAMTS13 plasma levels during the third trimester,49 which are more severe when obstetric complications such as HELLP syndrome occur.50 However, most pregnancies following recovery are uncomplicated.47 Inflammatory conditions, such as pancreatitis,51 infections,52 and surgery,53 may also trigger acute episodes of TTP.
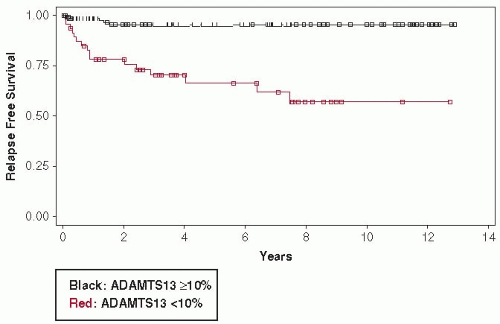
FIGURE 99.2 Kaplan-Meyer analysis of time to relapse for patients with ADAMTS13 activity <10% and patients with ADAMTS13 activity ≥10% at the time of their initial diagnosis of TTP. Data are from all 47 surviving patients who had initially presented with ADAMTS13 activity <10% and all 136 surviving patients with ADAMTS13 activity ≥10% who presented to the Oklahoma TTP-HUS Registry, 1995 to 2008. Median follow-up of all 47 patients with ADAMTS13 activity <10% was 7.5 years; median follow-up of 26 living patients with ADAMTS13 activity <10% who had not relapsed was 4.6 years. (Adapted from Kremer Hovinga JA, Vesely SK, Terrell DR, et al. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood 2010;115:1500-1511.)
Clinical and Laboratory Features
Validity of the minimal diagnostic criteria of microangiopathic hemolytic anemia and thrombocytopenia, in the absence of another clinically apparent cause,6,15 is supported by the presenting features of patients who have TTP associated with acquired, severe ADAMTS13 deficiency. Many of these patients have no neurologic or renal function abnormalities.6 The occurrence of the “classic pentad” of diagnostic features described nearly 50 years ago13 now rarely occurs, and then only in atypical patients6 see Table 99.3. Serum LDH levels are typically very high, reflecting severe hemolysis and also the ischemic injury of multiple organs.54 Case reports have described three patients who had recovered from TTP associated with severe ADAMTS13 deficiency and subsequently had stroke symptoms that were attributed to TTP even in the absence of thrombocytopenia and microangiopathic hemolytic anemia.55,56 The role of TTP as the etiology of the stroke symptoms was postulated because of the presence of severe ADAMTS13 deficiency and apparent response to plasma exchange treatment.55,56 The occurrence of neurologic abnormalities in the absence of microangiopathic hemolytic anemia and thrombocytopenia is probably rare, but the possibility is an important clinical concern.
Only gold members can continue reading. Log In or Register to continue
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Inherited Thrombocytopenias
Inherited Thrombocytopenias
 Unusual Sites of Arterial Occlusion
Unusual Sites of Arterial Occlusion
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient



