The Skeletal System in Hypothyroidism
Graham R. Williams
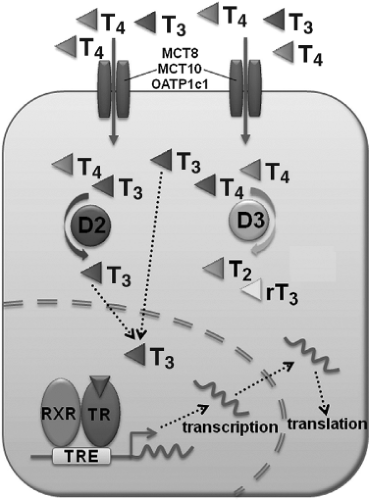 Figure 42.1 Mechanism of thyroid hormone action. Entry of T4 and T3 into target cells is mediated by monocarboxylate transporters MCT8 and MCT10, and the OATP1c1. The relative activities of the type 2 and type 3 deiodinase enzymes (D2, D3) regulate the intracellular availability of the active thyroid hormone T3. D2 converts the pro-hormone T4 to T3, whereas D3 inactivates both T4 and T3 converting them to T2 and reverse T3 (rT3). T3 enters the nucleus and binds the nuclear T3 receptor (TR) with high affinity. The hormone-bound receptor interacts with retinoid X receptor (RXR) and the TR/RXR heterodimer complex interacts with thyroid hormone response element sequences (TRE) located in the promoter regions of T3 target genes to control their expression. Reprinted from Gogakos AI, Duncan Bassett JH, Williams GR. Thyroid and bone. Arch Biochem Biophys 2010;503(1):129–136, with permission from Elsevier. |
Physiologic Control of Thyroid Hormone Action
The hypothalamic–pituitary–thyroid (HPT) negative feedback axis regulates circulating concentrations of thyroxine (T4), 3,5,3′-L-triiodothyronine (T3), and thyrotropin (thyroid-stimulating hormone, TSH), thereby controlling physiologic thyroid status (1). The main hormone secreted by the thyroid gland is T4, which enters peripheral tissues by active transport along with smaller amounts of T3 (2) (Fig. 42.1). T4 and T3 are then metabolized by the type 1, type 2, and type 3 iodothyronine deiodinase enzymes (Dio1, Dio2, and Dio3). Dio1 and Dio2 activate T4 by catalyzing removal of a 5′-iodine atom to generate the active hormone T3, whereas Dio3 prevents activation of T4 and inactivates T3 by removal of a 5-iodine atom (3). Although the physiologic role of Dio1 is incompletely understood, the enzyme is expressed principally in liver and kidney and is a major contributor to the circulating pool of T3. The physiologic importance of the Dio2 and Dio3 enzymes in hormone responsive T3-target cells, however, is more clearly defined. For example, Dio3 activity is maintained at high levels in embryonic tissue but falls rapidly at birth. By contrast, Dio2 activity is low in the fetus and rises following birth, and thus the coordinated and reciprocal expression of Dio3 and Dio2 enzyme activities represents a developmental switch that maintains cell proliferation during embryogenesis and then triggers T3-induced cell differentiation and organogenesis in a variety of tissues. Pre-receptor control of T3 availability in hormone responsive target cells, therefore, represents a key mechanism that regulates thyroid hormone action in a tissue specific manner (4,5,6,7,8).
Thyroid hormone actions are mediated ultimately by TRα1 and TRβ1 nuclear receptors, which bind T3 with high affinity and function as hormone-activated transcription factors (9) (Fig. 42.1). In the absence of hormone, unliganded TRs mediate transcriptional repression of target genes, whilst T3 binding stimulates hormone-dependent activation of gene transcription. TRα1 and TRβ1 are expressed widely but in varying concentrations and at different times in individual tissues (10). Together with the pre-receptor control of T3 availability this is a powerful mechanism that controls coordinated temporal and spatial responses to thyroid hormones in key target organs, including the skeleton, during development and in adulthood.
Thyroid Hormone Metabolism in Bone
The thyroid hormone transporter protein monocarboxylate transporter-8 (MCT8) is expressed in growth plate chondrocytes, bone-forming osteoblasts, and bone-resorbing osteoclasts at all stages of cell differentiation (11), although its functional importance has not been determined. Recent studies indicate that organic acid transporter protein-1c1 (OATP1c1) and another membrane transporter MCT10 also facilitate thyroid hormone uptake in target tissues (2). OATP1c1 mRNA is
not expressed in osteoblastic cells (12), but its expression in other bone cell lineages has not been investigated and it is also not known whether MCT10 is present in bone or cartilage.
not expressed in osteoblastic cells (12), but its expression in other bone cell lineages has not been investigated and it is also not known whether MCT10 is present in bone or cartilage.
The T3 and T4 inactivating enzyme, Dio3, is expressed widely in the skeleton and the highest levels of activity occur in growth plate chondrocytes prior to weaning (11). Studies in developing mice indicate that high levels of Dio3 are present in fetal bone until the time of birth when Dio3 expression falls. At this time, Dio2 activity rises suggesting that reciprocal coordination of Dio3 and Dio2 expression facilitates increased availability of T3 during the critical postnatal period when thyroid hormones are essential to promote growth (13). Studies of mice lacking Dio1 or with deficiencies of both Dio1 and Dio2 revealed that linear growth was not compromised (see later) (14,15,16,17). Several laboratories subsequently showed that Dio1 is not expressed in bone and cartilage (11,18,19,20), indicating that the enzyme does not influence T3 action in the skeleton directly.
In contrast, Dio2 plays an important role to regulate T3 availability both during skeletogenesis and in adult bone. Dio2 activity in the perichondrium surrounding the embryonic chick growth plate is involved in regulation of the rate of chondrocyte differentiation during early bone formation and growth (5), while increased activity of the enzyme during late embryogenesis in mice indicates its importance to control T3 responsiveness in developing bone in the early postnatal period (13). Studies of knockout mice lacking Dio2, however, demonstrate that deletion of Dio2 does not affect linear growth in juveniles. Furthermore, adult knockout mice are of normal size, suggesting that the importance of Dio2 in development (5,13) may be restricted to the immediate postnatal period and that other pathways compensate to ensure that growth is maintained later on during adolescence (21). Despite the presence of normal skeletal development and growth in Dio2 knockout mice, adult animals had brittle bones with impaired resistance to fracture. This phenotype was due to an isolated reduction of osteoblastic bone formation that resulted in a generalized increase in bone mineralization density (BMD) (21). Intracellular T3 deficiency was restricted to osteoblasts, indicating that maintenance of adult bone mineralization and optimal bone strength requires adequate supplies of T3 in osteoblasts that are generated by Dio2 (21). The findings suggest that restricted expression of Dio2 in adult bone (11) is necessary to maintain a higher intracellular T3 concentration in osteoblasts relative to other skeletal cells. As in other tissues, Dio2 activity in osteoblasts increases in hypothyroidism and is reduced in hyperthyroidism (19), suggesting that Dio2 buffers the effects of altered serum thyroid hormone levels on the skeleton by maintaining optimal intracellular T3 concentrations in osteoblasts to mitigate adverse effects of thyroid hormone deficiency or excess on bone mineralization and strength (21).
Thyroid Hormone Receptor Function in Bone
Both TRα1 and TRβ1 are expressed in bone and quantitative RT–PCR studies reveal that levels of TRα1 are at least 10-fold greater than levels of TRβ1 (22,23), suggesting that TRα1 is the primary mediator of T3 action in skeletal cells. TRα1 and TRβ1 expression has been documented clearly in growth plate chondrocytes, bone marrow stromal cells, and osteoblasts, but it remains uncertain whether TRs are expressed in osteocytes or osteoclasts (24,25). The assumption that TRα1 is the primary mediator of T3 action in skeletal cells is supported by numerous studies in mice with mutation or deletion of the Thra or Thrb genes. Thus, mutation or deletion of TRα results in a striking skeletal phenotype that is distinct and opposite to the abnormalities observed in mice with mutation or deletion of TRβ. Overall, mice with mutation or knockout of TRα are euthyroid but display developmental abnormalities that are characteristic of hypothyroidism, including delayed endochondral ossification, disorganised growth plate architecture, impaired chondrocyte differentiation, growth retardation, and reduced bone mineral deposition during growth (25,26,27,28,29). Adult mice with mutation or deletion of TRα have osteosclerosis with increased bone mineralization (23,25,26,27,29,30), consistent with reduced bone turnover resulting from impaired thyroid hormone action in skeletal cells during adulthood (31). Mice with mutation or knockout of TRβ, by contrast, have elevated thyroid hormone and TSH concentrations due to impaired negative feedback control of the HPT axis, thus revealing resistance to thyroid hormone (RTH) in the pituitary and hypothalamus. The elevated circulating thyroid hormone concentrations in TRβ mutant mice result in accelerated endochondral and intramembranous ossification with advanced bone age resulting in increased bone mineral content (23,27,29). Growth is accelerated early in the postnatal period but ultimately there is short stature due to premature quiescence of the growth plates. In adults with mutation or deletion of TRβ there is osteoporosis with increased bone resorption (23,25,27,29,30), features that are typical of thyrotoxicosis and consistent with increased bone turnover and bone loss due to excess thyroid hormone signaling in adult bone (31). Studies in TR mutant mice thus provide compelling evidence that TRα is the major mediator of thyroid hormone action in bone and that the skeletal consequences of mutation or knockout of TRβ are secondary to disruption of the HPT axis and raised circulating thyroid hormone concentrations (25,30). Nevertheless, TRβ is expressed in bone and recent studies suggest this isoform may also be functional during endochondral ossification (32) and in the adult skeleton (33), although further studies are required to establish the physiologic importance of TRβ in normal bone development and maintenance.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


