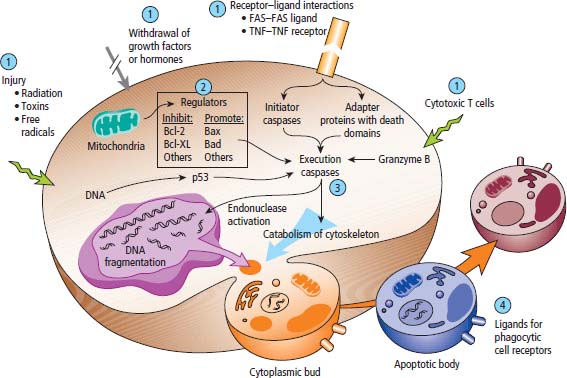
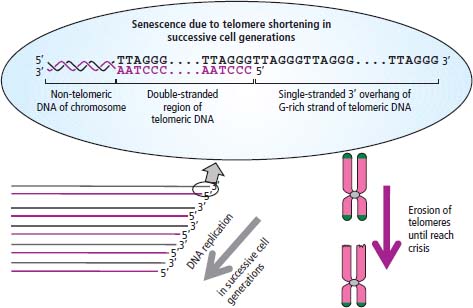
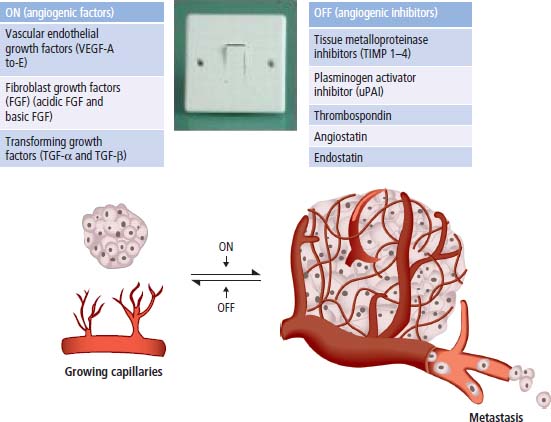
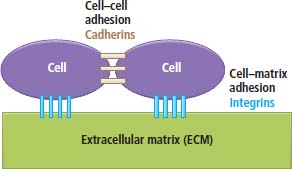
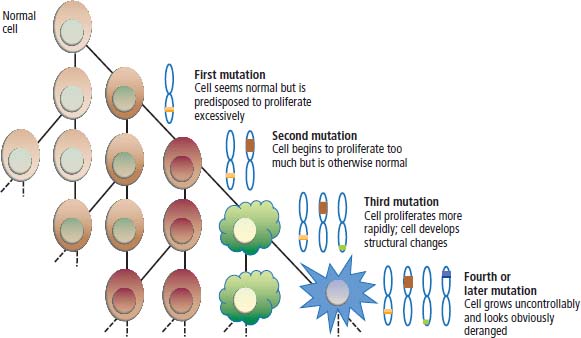
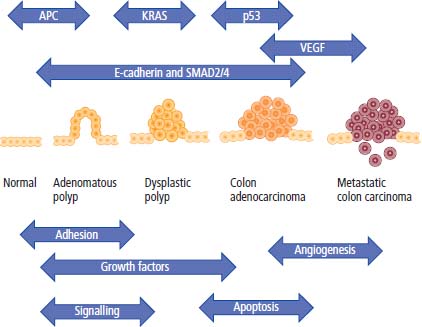
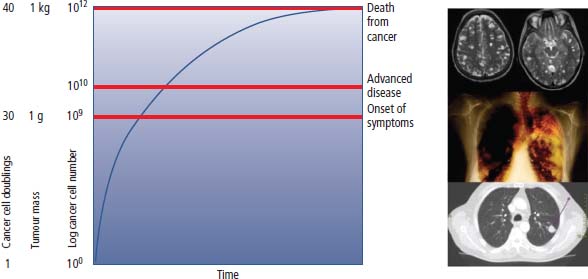
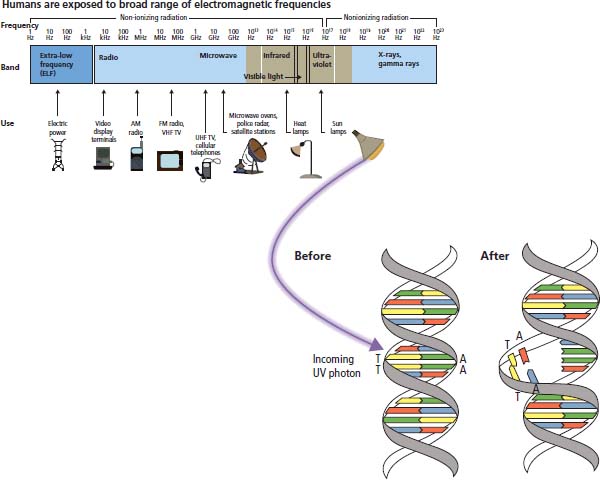
2 At a molecular level, six basic steps or “hallmarks” that turn a cell into a cancer were described in 2000. In 2011, a further two “enabling hallmarks” were added that contribute to the ability of cells to acquire the six hallmarks and a further two “emerging hallmarks” required for cancer cells to continue to survive as tumours. The original six hallmarks of cancer are: It is probable that all six are necessary to a greater or lesser extent. Some molecular changes may contribute to more than one of the six capabilities (e.g. a mutation on the p53 gene may contribute to both avoidance of apoptosis and insensitivity to inhibitory stimuli). A number of mechanisms may contribute to the acquisition of these six properties, including genomic instability as a consequence of deficient DNA repair (lack of caretaking) or loss of cell cycle arrest/death in response to DNA damage (lack of gatekeeping). Many new treatment strategies for cancer aim to interrupt these steps. The two enabling hallmarks of cancer are: The two emerging hallmarks of cancer are: Figure 2.1 Signal transduction pathway. The instructions to cells to grow and start dividing are transmitted to cells by extracellular growth factor ligands that bind cell surface receptors. This results in the reversible phosphorylation of tyrosine, threonine or serine residues. The transfer of these molecular switches from activated receptors to downstream nuclear transcription activators is known as signal transduction (Figure 2.1). This cascade results in amplification of the initial stimulus. Cancers achieve self-sufficiency in growth factors and are not dependent on extracellular concentrations of growth factors for continued growth. The majority of dominant oncogenes act on this mechanism by one of the following mechanisms: Many normal cells grow throughout their lifespan and the co-ordination of their growth, differentiation, senescence and death is controlled by the cell cycle. Antiproliferative signals may be received by cells as soluble growth inhibitors or fixed inhibitors in the extracellular matrix. They act on the cell cycle clock (Box 2.1 and Figure 2.3), most frequently arresting transit through G1 into the S phase. Cancer cells ignore these “STOP” signals. The co-ordination of the cell cycle and its arrest at checkpoints in response to DNA damage is achieved by sequential activation of kinase enzymes that ultimately phosphorylate and dephosphorylate the retinoblastoma protein (Rb). Periodic activation of these cyclin–cyclin-dependent kinase (CDK) complexes drives the cell cycle forward (Figure 2.2). Phosphorylation of Rb releases E2F, a transcription factor, which is then able to promote the expression of a number of target genes resulting in cell proliferation. The brakes that balance this system are CDK inhibitors (CKIs). Interference in elements of the cell cycle regulatory process is a common theme in malignancy (Table 2.1). Examples of independence of cell cycle checkpoints: Figure 2.2 Oscillating levels of cyclins through the phases of the cell cycle. An important checkpoint or restriction point in the cell cycle occurs in G1 to ensure that errors in DNA are not replicated, but instead are either repaired or that the cell dies by apoptosis. This is initiated by damaged DNA and is co-ordinated by p53, the gene that is probably most commonly mutated in cancers overall. Additional checkpoints are present in the S and G2 phases to allow cells to repair errors that occur during DNA duplication and thus prevent the propagation of these errors to daughter cells. Table 2.1 Examples of the six features and their molecular basis in cancers APC, adenomatous polyposis coli gene; EGFR, endothelial growth factor receptor; hTERT, human telomerase reverse transcriptase; MMP, matrix metalloproteinase; PDGF, platelet-derived growth factor; PDGFR, platelet-derived growth factor receptor; VEGF, vascular endothelial growth factor; UPA, urokinase-type plasminogen activator; SMAD, small mothers against decapentaplegic; CDK, cyclin-dependent kinase; KRAS, Kirsten rat sacroma viral oncogene. Apoptosis is a pre-programmed sequence of cell suicide that occurs over 30–120 minutes. Apoptosis commences with condensation of cellular organelles and swelling of the endoplasmic reticulum. The plasma membrane remains intact, but the cell breaks up into several membrane-bound apoptotic bodies, which are phagocytosed. Confining the process within the cell membrane reduces activation of both inflammatory and immune responses, so that programmed cell death does not cause autoimmune disease or inflammation. Amongst the molecules that control apoptosis are the Bcl-2 family that confusingly includes both pro-apoptosis members (e.g. Bax) and anti-apoptosis members (e.g. Bcl-2). In mammalian cells two pathways initiate apoptosis (Figure 2.4): Figure 2.3 The cell cycle. Ultimately both pathways activate the caspase cascade, a series of protease enzymes that result in cell apoptosis. Evasion of this pathway is a prerequisite for malignant cell proliferation and a number of strategies to this end have been identified (Table 2.1). In culture, cells can divide a limited number of times, up to the “Hayflick limit” (60–70 doublings in the case of human cells in culture), before the cell population enters crisis and dies off. This senescence is attributed to progressive telomere loss, which acts as a mitotic clock (Figure 2.5). Telomeres are the end segments of chromosomes and are made up of thousands of copies of a short six-base pair sequence (TTAGGG). Figure 2.4 The apoptotic pathway. Figure 2.5 Telomerase, telomere length, senescence and immortalization. DNA replication always follows a 5′ to 3′ direction so that manufacturing the 3′ ends of the chromosomes cannot be achieved by DNA polymerases and each time a cell replicates its DNA ready for cell division, 50–100 base pairs are lost from the ends of chromosomes. Eventually the protective ends of chromosomes are eroded and end-to-end chromosomal fusions occur with karyotypic abnormalities and death of the affected cell. Normal germ cells and cancer cells avoid this senescence, acquiring immortality in culture usually by upregulating the expression of human telomerase reverse transcriptase (hTERT) enzyme that uses an RNA template and RNA-dependent DNA polymerase to add the six-base pair sequence back onto the ends of chromosomes to compensate for the bases lost during DNA replication (Table 2.1). Dyskeratosis congenita is an inherited condition, characterized by many abnormalities, including premature ageing and an increased risk of skin and gut cancers. It is due to mutations of components of the telomerase complex including the telomerase RNA and dyskerin. Figure 2.6 The angiogenic switch. All tissues including cancers require a supply of oxygen and nutrients. For cancers to grow larger than about 0.4 mm in diameter, a new blood supply is needed to deliver these. The growth of new blood vessels from pre-existing vasculature is termed angiogenesis. The “angiogenic switch” denotes the ability of tumours to recruit new blood vessels by producing growth factors and is necessary for tumour growth and metastasis. Angiogenesis is determined by the balance of angiogenesis promoters and inhibitors (Figure 2.6). Vascular endothelial growth factors (VEGF-A to -E) are a family of growth factor homodimers that act via one of three plasma membrane receptors (VEGFR-1 to -3) on endothelial cells. Overproduction of VEGF and/or FGF (fibroblast growth factor) is a common theme in many tumours (Table 2.1). Angiogenesis may be measured microscopically as microvessel density in an area of tumour or by assays of angiogenic factors. These measures are of prognostic significance in several human tumours. Angiogenesis is becoming a major focus of anticancer drug development. It is an attractive target for several reasons. Angiogenesis is a normal process in growth and development but is quiescent in adult life except during wound healing and menstruation, so side effects are predicted to be minimal. As the target will be normal endothelial cells without any genetic instability, there should be little capacity to acquire resistance. Each capillary supplies a large number of tumour cells, so the effects should be magnified in terms of tumour cell kill. Anti-angiogenic agents should have easy access to their target through the blood stream. In combination, these elements make anti-angiogenic therapies attractive, and several pharmaceutical companies have invested heavily in attempts to develop these agents. Bevacizumab is a monoclonal antibody that binds to VEGF; it was originally licensed for use in colon cancer and is also a valuable treatment for age-related macular degeneration, which is caused by retinal vessel proliferation. Examples of neoangiogenesis in cancers: The properties of tissue invasion and metastatic spread are the histopathological hallmarks of malignant cancers that discriminate them from benign. A number of sequential steps have been identified in the process of metastatic spread of cancers: Central to many of these steps is the role of cell–cell adhesion that controls the contact between cells and cell–extracellular matrix connections that influence the relationship between a cell and its environment. These interactions are regulated by cell adhesion molecules. Members of the cadherin and immunoglobulin superfamilies modulate cell–cell interactions whilst integrins control cell–extracellular matrix interactions. Alterations of cadherin, adhesion molecule and integrin expression are a common feature of metastatic cancer cells (Table 2.1). Tumours may migrate as single cells or as collections of cells. The former strategy is used by lymphoma and small-cell lung cancer cells. It requires changes in integrins that mediate the cell–extracellular matrix interaction and matrix-degrading proteases. Metastatic migration as clumps of cancer cells is common for most epithelial tumours. In addition, this needs changes in cell–cell adhesion through cadherins and other adhesion receptors, as well as cell–cell communication via gap junctions (Figure 2.7). Figure 2.7 Cell–cell and cell–matrix interactions. Cancer is really a genetic disease caused by mutations of cellular DNA that do not occur in the germ cells (oocytes and sperm). One of our lines of defense against cancer is to repair errors in DNA or to eradicate cells that have accumulated extensive DNA damage. However, DNA mutations tend to go uncorrected in cancer cells because their DNA replication is error-prone, their DNA repair mechanisms are deficient and their DNA damage cell cycle arrest and apoptosis responses are uncoupled. Altogether cancer cells are said to have genetic instability and this enables them to acquire the six hallmarks of a cancer cell. Sometimes these mutations develop in a stepwise fashion as the cell phenotype becomes more abnormal. Although cancers generally have ways of avoiding destruction by the host immune system (see hallmark 9. Cancers avoid destruction by the host immune system), they still often induce a local inflammatory response. It was thought that this inflammatory response was part of the host’s attempt to eradicate the cancer cells. Instead it appears that the inflammatory cytokines actually promote tumour growth, proliferation and angiogenesis. Rather like autoimmune disease it seems that this is another own goal of the immune system. Cancers employ a number of strategies to escape the host immune system so that they can survive and grow. They have acquired ways of avoiding both the innate and adoptive immune systems and this is why for most cancers, immunotherapy is ineffective. Cancer cell proliferation is enabled by changes in energy metabolism to fuel growth. In many circumstances cancer cells switch from normal aerobic glycolysis to the much less efficient anaerobic metabolism. Although the rationale behind this metabolic switch in cancer cells is unclear, it may account for the resistance of some tumours to the effects of radiotherapy. DNA damage or mutation will normally result in cell cycle arrest followed by DNA repair or apoptosis. Interference in this process may occur either by deficient DNA damage recognition and repair or abnormal gatekeeping of the cell cycle arrest/apoptosis response. This will result in the uncorrected accumulation of a large number of genetic abnormalities, which is referred to as “genomic instability”. It is thought that this allows cells to acquire the six capabilities that characterize the cancer cell phenotype and physiology (Figures 2.8 and 2.9). Environmental damage to DNA occurs commonly and eukaryotes have developed several techniques for repairing both double strand breakages and single strand errors in DNA. Figure 2.8 Stepwise accumulation of genetic mutations contributing to oncogenic phenotype. Figure 2.9 Colon cancer development from normal mucosa to metastatic carcinoma associated with stepwise acquisition of oncogenic mutations. APC, adenomatous polyposis coli gene; KRAS, Kirsten rat sarcoma viral oncogene homologue; p53, tumour protein 53 (TP53); SMAD, Homo sapiens homologue of Drosophila protein mothers against decapentaplegic (MAD); VEGF, vascular endothelial growth factor. Hereditary mutations of the enzymes involved in DNA repair will predispose to malignancy as they confer genome instability (Table 2.2). Table 2.2 Hereditary DNA repair syndromes Another group of enzymes are required to recognize damaged DNA, leading to cell cycle arrest to allow DNA repair to be completed before the damage is replicated and passed on to the progeny cells. A number of cancer-predisposing syndromes are associated with inherited mutations of these enzymes. Examples include p53, whose inactivation is an early step in the development of many cancers. Patients with the Li–Fraumeni syndrome carry one mutant germline p53 allele and are at high risk for the development of sarcomas, leukaemia and cancers of the breast, brain and adrenal glands. Most of the discussion above about the molecular mechanisms of malignancy has described somatic and occasional germline mutations of DNA that lead to aberrant proteins that in turn contribute to oncogenesis. This argument follows the central dogma of molecular biology introduced by Francis Crick in the late 1950s, which stated that information flows in a unidirectional course from DNA sequence via RNA sequence to protein sequence. Although there are recognized exceptions to the central dogma, such as retroviruses and prions, it remains broadly true. However, some inheritable changes in phenotype or gene expression arise by mechanisms other than changes in the sequence of DNA bases. These inheritable changes passed on from a cell to her daughters are called epigenetic changes and perhaps the most obvious of these is cell differentiation. The term epigenetics was introduced by the British developmental biologist Conrad Hal Waddington in 1942 as a metaphor for cell differentiation and development from a progenitor stem cell. Waddington likened differentiation to a marble rolling down a landscape of hills and valleys to reach a final destination. The destination (cell fate) was determined by the landscape (epigenetics) and the marble could not travel back to the top (terminal differentiation). Today the term refers to modification of DNA and chromatin that influences gene transcription, alteration of post-transcriptional RNA and finally to protein degradation. Perhaps the most recognized epigenetic modification of DNA is nucleotide base methylation, typically the addition of a methyl group to the cytosine pyrimidine ring. In vertebrates, DNA methylation usually occurs in a CpG dinucleotide. Unmethylated CpGs are grouped in clusters called “CpG islands” that occur in the 5′ regulatory regions of many genes. DNA methylation of CpG islands inhibits gene transcription by impeding the binding of transcriptional proteins and by binding methyl-CpG-binding domain (MBD) proteins. MBD proteins recruit additional proteins, such as histone deacetylases (HDACs), which modify histones to form compact, inactive chromatin termed silent chromatin. Since epigenetic changes such as DNA methylation are inherited during cell replication, maintenance of the pattern of DNA methylation is required following each cycle of DNA replication and this is achieved by DNA methyltransferases using the conserved DNA strand as the template (Figure 2.10). DNA methylation of tumour suppressor genes has been found to be a common mechanism of epigenetic gene silencing in cancers. Figure 2.10 DNA methylation is passed on during cell replication to progeny cells by DNA methyltransferase enzymes that methylate CpG islands. CpG refers to the DNA dinucleotide sequence CG joined by the phosphate backbone of DNA. Chromatin is composed of DNA and proteins, chiefly the histone proteins around which the DNA is wound. There are six classes of histones organized into core histones (H2A, H2B, H3 and H4) and linker histones (H1 and H5). The core histones, which are highly conserved through nature, share N-terminal amino acid sequences that are the sites for post-transcriptional modification, for example, acetylation and methylation. These histone modifications alter the binding of the DNA to the nucleosome and modify RNA polymerase activity and hence gene expression. In general, tightly bound DNA is less expressed. Numerous enzymes have been identified that are involved with the modification of histone protein leading to alterations of chromatin structure and regulation of gene expression. These include histone methyltransferase (HMT) and histone acetyltransferase (HAT); other enzymes catalyze the removal of these modifications including HDAC (Figure 2.11). Acetylation of histone tails reduces their binding affinity for DNA, allowing access for RNA polymerase and enhancing gene transcription. HDAC, therefore, by reversing histone tail residue acetylation suppresses gene expression, including tumour suppressor gene expression contributing to oncogenesis. A number of HDAC inhibitors have been studied including valproate and more recently vorinostat or suberoylanilide hydroxamic acid (SAHA), which is licensed for the management of cutaneous T-cell non-Hodgkin’s lymphoma. Post-translational interference of messenger RNA (mRNA) transcripts can also modify the expression of genes without altering the DNA sequence. Two types of small RNA molecules, microRNA (miRNA) and small interfering RNA (siRNA), can bind to specific complementary sequences of RNA or DNA and either increase or decrease their activity, for example by preventing an mRNA from producing a protein (Figure 2.12). RNA interference was originally identified in petunia plants. Botanists attempting to produce darker and darker petunia flowers inserted additional genes of an enzyme that catalyzes pigment synthesis. However, the transgenic plants produced white or variegated white flowers and this was subsequently found to be due to post-transcriptional inhibition of gene expression brought about by rapid mRNA degradation. The eventual explanation of this gene silencing phenomenon was identified in Caenorhabditis elegans by Craig Mello and Andrew Fire in 1998 who demonstrated that double-stranded RNA caused the gene silencing. They called this RNA interference (RNAi) and won the Nobel Prize in 2006 for this work. Both the role of RNAi in the epigenetic generation of cancers and the potential of RNAi as a therapeutic approach are the focus of fevered research. A further form of epigenetic modification that contributes to the cellular phenotype is the destruction of proteins chiefly by proteasomes. Proteins are tagged for degradation by a small protein called ubiquitin and this reaction is catalyzed by enzymes including the product of the gene disrupted in Von Hippel–Lindau syndrome and Fanconi’s anaemia. At least four ubiquitin molecules attach to the condemned protein in a process called polyubiquitination and the protein then moves to a proteasome, where the proteolysis occurs (Figure 2.13). Epigenetic regulation of protein degradation could contribute to oncogenesis in a variety of ways. Gankyrin, a component of the proteasome, is overexpressed in hepatocellular cancers. Bortezomib, a new treatment for myeloma, acts by inhibiting proteasome function. Figure 2.11 Mechanism of chromatin modification. Ac, acetyl; HAT, histone acetyltransferase; HDAC, histone deacetylase; HMT, histone methyltransferase; Me, methyl. Figure 2.12 Mechanism of RNA interference. The causes of cancer may be usefully divided into genetic and environmental factors. The genetic factors are either germline mutations that are present in every cell of the body or somatic alterations only found in the tumour cells. Germline mutations may be either inherited, in which case they follow a familial pattern or may be new sporadic mutations that neither parent has. Some of the germline mutations have been outlined as mutator phenotypes (DNA repair and damage recognition genes) above. Other germline cancer-predisposing mutations occur in tumour suppressor genes and oncogenes. The first clue to the identification of specific genes involved in the development of cancer came from the study of tumour viruses. Although cancer is generally not an infectious disease, some animal leukaemias, lymphomas and solid tumours, particularly sarcomas, can be caused by viruses. Oncogenes were identified following the discovery by Peyton Rous in 1911 that sarcomas could be induced in healthy chickens by injecting them with a cell-free extract of the tumour of a sick chicken. This was due to transmission of Rous sarcoma virus (RSV), an oncogenic retrovirus with just four genes: It is the src gene that is necessary for cell transformation and is therefore an oncogene – literally a gene capable of causing cancer. In the late 1970s Harold Varmus and Michael Bishop discovered that a homologous proto-oncogene (c-SRC) is present in the normal mammalian genome (the human src locus is on chromosome 20q12-q13) and has been hijacked by the retrovirus. The prefix v- denotes a viral sequence and the prefix c- a cellular sequence. In 1956, 55 years after his discovery of RSV and at the age of 87, Peyton Rous was (finally) awarded a Nobel Prize, whilst Bishop and Varmus only waited 10 years from their discovery to the award of their Nobel Prize in 1989. Around 50 oncogenes have been identified by their presence in transforming retroviruses (e.g. erbB, H-RAS, JUN) and further oncogenes have been discovered by positional cloning of chromosomal translocations (e.g. Bcl-2, BCR-ABL) and by transfection studies (e.g. N-RAS, RET). Most oncogenes contribute to cancer’s autonomy in growth factors, either as plasma membrane receptors (e.g. RET, PTCH), signal transduction pathways (e.g. PTEN, NF1 and 2, VHL) or transcription factors (e.g. c-MYC, WT1) (Table 2.3). Figure 2.13 Proteasome pathway of protein ubiquitination and degradation. E, ubiquitination enzymes; Ub, ubiquitin. In contrast to oncogenes, tumour suppressor genes act as cell cycle brakes, slowing the proliferation of cells, and mutations in these genes also contribute to cancer. Germline mutations of tumour suppressor genes behave as autosomal-dominant familial cancer predispositions. Tumour suppressor genes require the loss of both functional alleles to support a cancer (unlike oncogenes where one mutant allele suffices). In 1971 Alfred Knudson proposed the two hit model of tumour suppression to account for the differences between familial and sporadic retinoblastoma in children. In familial cases, tumours arose at a younger age and were more frequently bilateral. Knudson hypothesized that these children had inherited one defective retinoblastoma gene allele, followed by loss of the function of the second allele in the cancer cells through a somatic mutation (Figures 2.14 and 2.15). Tumour suppressor genes, like oncogenes, also involve a variety of functional categories, including cell cycle regulation (e.g. p53, Rb), DNA repair and maintenance (e.g. BRCA1 and 2, MLH1, MSH2), as well as signal transduction (e.g. NF1, PTEN) and cell adhesion (e.g. APC) (Table 2.2). The Maths of Cancer or “How long have I had it?” (Figure 2.16) Table 2.3 Table of hereditary cancer predisposition syndromes AD, autosomal dominant; AR, autosomal recessive; GTPase, guanosine triphosphatase. The cell cycle takes 16–24 hours to complete. For Burkitt’s lymphoma the growth fraction is 0.29 (i.e. 29% of the cells are actively dividing at any one time) and hence the doubling time is about 2 days. In contrast, the growth fraction for non-small cell cancer of the lung is under 0.02 and the doubling time is around 130 days. To get to death (1012 cells) can take just 3 months for BL but for NSCLC to get to 109 (just detectable on CT) takes 10.5 years. In reality most lung cancers are found when they are 3 cm in diameter or 1010.5 cells, after 35 doublings (12.3 years). The multitude of environmental factors that are associated with the development of malignancy may be usefully divided into: Figure 2.14 Knudson’s two-hit hypotheses of familial and sporadic retinoblastoma. The major physical carcinogen is radiation. Radiation is ubiquitous and may either be ionizing (e.g. γ-rays from cosmic radiation and isotope decay, α-particles from radon, X-rays from medical imaging) or non-ionizing (e.g. ultraviolet (UV) light from the sun, microwave and radiofrequency radiation from mobile phones, electromagnetic fields from electricity generators and pylons, ultrasound radiation from imaging). Ionizing radiation ejects electrons from atoms yielding an ion pair and requires 10–15 eV (electronvolts). Ionizing radiation may be either electromagnetic (X-rays, γ-rays) or particulate (α-particles, neutrons). Non-ionizing radiation does not yield an ion pair but can still excite electrons resulting in chemical change. Figure 2.15 Multiple dermal neurofibromata typical of peripheral neurofibromatosis or type 1 NF, previously known eponymously as von Recklinghausen’s disease. It is due to hereditary mutation of the NF1 neurofibromin gene on chromosome 2p22, which encodes a guanosine triphosphatase (GTPase) activating protein involved in the signal transduction cascade. UV radiation is electromagnetic radiation with a wavelength shorter than visible light (400–700 nm) but longer than X-rays (10–0.01 nm). It is subdivided into three wavelength bands: UVC has the most potent effects on DNA, which absorbs most strongly at 254 nm. However, UVC is quickly absorbed by the atmosphere and hence UVB is considered to be the greater environmental hazard. Most UV radiation is absorbed by atmospheric ozone in the stratosphere and this ozone layer is being depleted in part due to chlorine in chlorofluorocarbons (CFCs), resulting in increasing UV exposure levels. Ultraviolet light is responsible for vitamin D activation and suntans. One of the major lesions induced in DNA by UV radiation is the thymidine dimer, a covalent bonding of adjacent thymidine residues on the same DNA strand (Figure 2.17). This causes local distortion of the double helix that is repaired by the NER pathway. The seven identified xeroderma pigmentosa genes encode essential components that undertake NER and hence xeroderma pigmentosa predisposes to UV-induced skin malignancies. Melanin pigment in the skin normally absorbs UV radiation, thus protecting the skin. Basal cell and squamous cell skin cancers increase with cumulative UV exposure, whilst the relationship is less straightforward for melanoma. The evidence for an association with cancer for other forms of non-ionizing radiation (microwave, radiofrequency, ultrasound and electromagnetic radiation) is weak and inconsistent. Figure 2.16 The maths of cancer. Figure 2.17 UV-light-induced thymidine dimers. T, Thymine; A, Adenine. Exposure to natural sources of ionizing radiation varies in different populations. Higher altitude and further latitude from the equator are both associated with higher cosmic radiation exposure composed of very high energy particles that are thought to originate outside our solar system including from supernovae. In addition, various regions have higher natural background levels from radon. Radon is a colourless, odourless gas formed from decay as part of the uranium-238 series. The radon-222 isotope, along with a number of its progeny, is an α-particle emitter. Radon gas levels are normally quoted in Bq/m3 (1 becquerel (Bq) is one decay per second) and the average indoor levels in the United Kingdom are about 20 Bq/m3. Local geology (igneous granite) with high levels of uranium produces high levels of radon in soil gas, but for it to escape to the surface the soil must be highly porous. In the United Kingdom, radon levels are particularly high in Devon and Cornwall, Derbyshire and Northamptonshire. From the results of eight case-control studies, it is believed that radon exposure accounts for a small fraction of lung cancers with a 14% increased risk for a person living for 30 years in a house with levels of 150 Bq/m3. Figure 2.18 How atomic bombs kill. Most of the information on the induction of cancers by ionizing radiation comes from exposed populations, including Japanese people exposed to atomic bombs at Hiroshima (“Little Boy” was a uranium-235-enriched bomb dropped by Enola Gay) and Nagasaki (“Fat Man” was a plutonium-239 bomb dropped by Bockstar). The estimated populations of the two cities at the time of bombing was 560,000 and approximately 200,000 people died within the first few months of the acute effects of blast, burns and radiation exposure (Figure 2.18). The Radiation Effects Research Foundation has followed 86,000 survivors or hibakusha and, up to 1990, 7827 had died of cancer. The excess risk of leukaemia was seen especially in those exposed as children and was highest during the first 10 years after the bombing. However, the excess risk of solid tumours occurred later and still persists (Table 2.4). Table 2.4 Cancer deaths in the hibakusha (survivors of Hiroshima and Nagasaki atomic bombs)
The scientific basis of cancer
The hallmarks of cancer
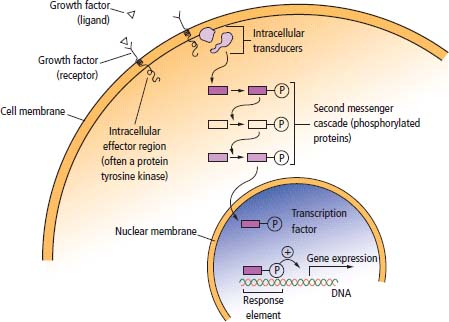
1. Sustaining proliferative signalling (growing by themselves)
2. Evading growth suppressors
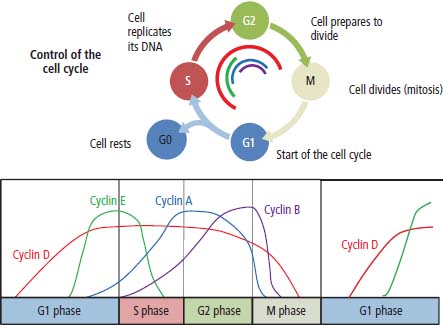
G1/S checkpoint
Feature
Colorectal cancer
Glioma
Head and neck squamous cancer
1. Growth factor independence
KRAS mutation
EGFR amplification or mutation NF1 loss
EGFR mutation
2. Over-riding inhibitory signals
SMAD2/SMAD4 mutation
CDK4/p16 mutation
Cyclin D amplification p16 and p21 mutation
3. Evasion of apoptosis
p53 mutation
p53 mutation/MDM2 overexpression
p53 mutation
4. Immortalization
hTERT re-expression
hTERT re-expression
hTERT re-expression
5. Angiogenesis
VEGF expression
PDGF/PDGFR overexpression
Nitric oxide pathway activation of VEGF
6. Invasion and metastasis
APC, inactivate E-cadherin
Cathepsin D, MMP-2 and -9 and UPA overexpression
Cathepsin D, MMP-1, -2 and -9 overexpression
3. Resisting cell death
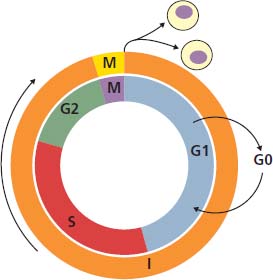
4. Enabling replicative immortality
5. Angiogenesis
6. Invasion and metastasis
Examples of invasion in cancers
How to acquire the six capabilities (enabling hallmarks)
7. Cancer cells are prone to mutations due to genome instability
8. Inflammation promotes the growth of cancers
Additional capabilities of cancers (emerging hallmarks)
9. Cancers avoid destruction by the host immune system
10. Cancer cells deregulate cellular metabolism
Genome instability
DNA repair
DNA damage
DNA repair mechanism
Examples of defect of DNA repair
Examples of cancers associated with defects
Double strand DNA breakage
Homologous (sister chromatid) repair
BRCA1 (hereditary breast and ovarian cancer)
Breast and ovarian cancers
Non-homologous end joining (NHEJ)
XRCC4 (X-ray repair complementing defect gene) (lethal)
None (lethal defect)
Single strand DNA breakage
Nucleotide excision repair (NER)
XP (xeroderma pigmentosa)
Skin cancers, leukaemia and melanoma
Mismatch repair (MMR)
MSH and MLH (hereditary non-polyposis colon cancer)
Colon, endometrium, ovarian, pancreatic and gastric cancers
Base excision repair (BER)
MYH (hereditary non-polyposis colon cancer)
Colon cancers
DNA damage recognition
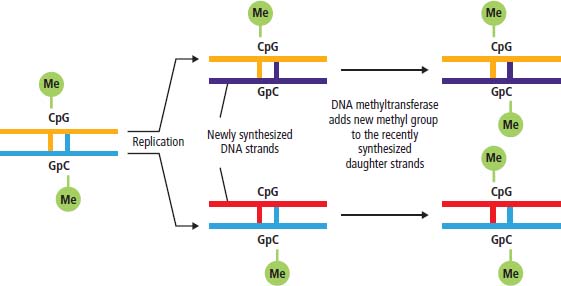
Epigenetic changes
DNA methylation
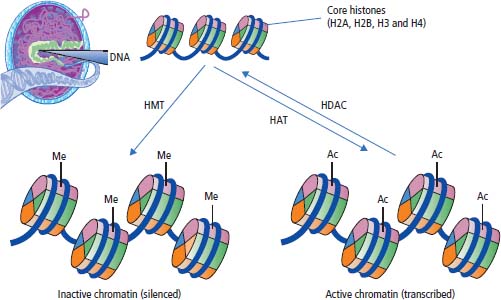
Chromatin modification
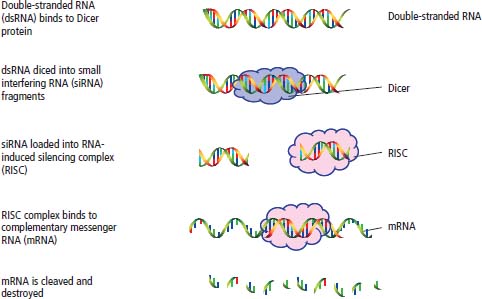
RNA interference
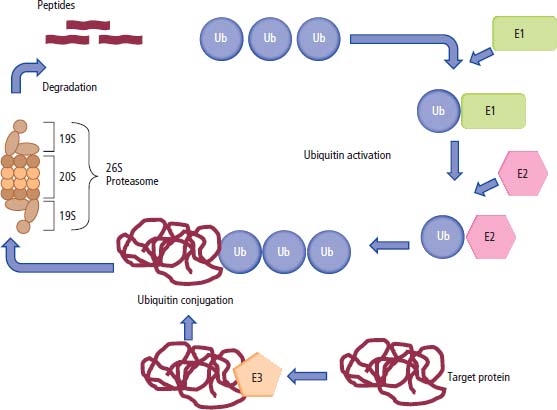
Protein degradation
Genetic causes of cancer
Hereditary causes of cancer
Oncogenes
Tumour suppressor genes
Syndrome
Malignancies
Inheritance
Gene
Function
Breast/ovarian
Breast, ovarian, colon, prostate
AD AD
BRCA1 BRCA2
Genome integrity
Cowden
Breast, thyroid, gastrointestinal, pancreas
AD
PTEN
Signal transduction (tyrosine phosphatase)
Li–Fraumeni
Sarcoma, breast, osteosarcoma, leukaemia, glioma, adrenocortical
AD
p53
Genome integrity
Familial polyposis coli
Colon, upper gastrointestinal
AD
APC
Cell adhesion
Hereditary non-polyposis colon cancer (Lynch type II)
Colon, endometrium, ovarian, pancreatic, gastric
AD AD AD AD
MSH2 MLH1 PMS1 PMS2
DNA mismatch repair
MEN 1 (multiple endocrine neoplasia 1)
Pancreatic islet cell, pituitary adenoma
AD
MEN1
Transcription repressor
MEN 2 (multiple endocrine neoplasia 2)
Medullary thyroid, phaeochromocytoma
AD
RET
Signal transduction (receptor tyrosine kinase)
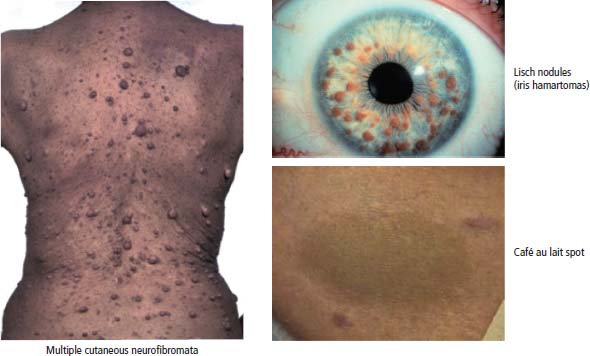
Neurofibromatosis 1 (Figure 2.15)
Neurofibrosarcoma, phaeochromocytoma, optic glioma
AD
NF1
Signal transduction (regulates GTPases)
Neurofibromatosis 2
Vestibular schwannoma
AD
NF2
Cell adhesion
von Hippel–Lindau
Haemangioblastoma of retina and central nervous system, renal cell, phaeochromocytoma
AD
VHL
Ubiquination
Retinoblastoma
Retinoblastoma, osteosarcoma
AD
RB1
Cell cycle regulation
Xeroderma pigmentosa
Skin, leukaemia, melanoma
AR AR AR AR
XPA XPC XPD XPF
DNA nucleotide excision repair
Gorlin
Basal cell skin, brain
AD
PTCH
Signal transduction (repressor of hedgehog signalling)
Environmental causes of cancer
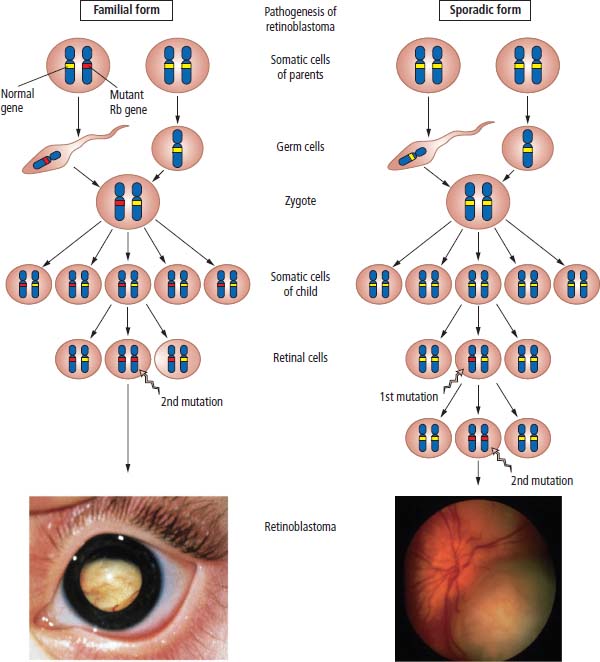
Radiation
Ultraviolet radiation
Ionizing radiation
Natural sources

Nuclear warfare
Total number of deaths
Estimated number of deaths due to radiation
Percentage of deaths attributable to radiation
Leukaemia
176
89
51
Solid tumours
4687
339
7
Total
4863
428
9
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


