THE RENIN-ANGIOTENSIN SYSTEM
ANGIOTENSINOGEN
Angiotensinogen (AGT), also termed renin substrate, is the precursor for the angiotensin peptides, which include angiotensin (A)-I, II, III, and IV, and A1–7. Levels of this peptide, which can be rate-limiting for the renin-angiotensin system, are produced mainly in the liver, where its precursor preproangiotensinogen is synthesized and glycosylated in the hepatocytes; nonetheless, there is evidence of production in other tissues as well (e.g., the heart, blood vessels, kidney, and adipocytes).
In the circulation, AGT, with a half-life of 16 hours, is cleaved by renin (˜10%) and/or other enzymes to release A-I (Fig. 79-1). In many tissues not expressing renin, AGT can be cleaved by enzymes other than renin (e.g., cathepsin G, tonin, and chymase). The extent to which AGT is glycosylated may influence the kinetics of renin in the circulation. Indeed, this has been hypothesized in a proposed, separate, brain-AGT system.
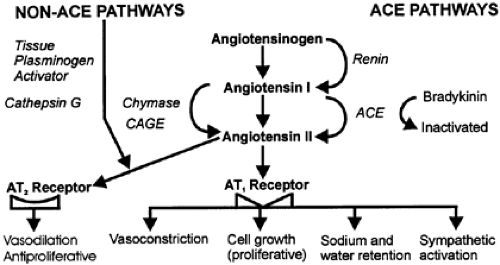 FIGURE 79-1. Angiotensinogen and angiotensin I and II pathways and their role in vascular and fluid homeostasis via angiotensin II receptors (AT 1 and AT 2). (CAGE, chymotrypsin-like angiotensin-generating enzyme.) |
There is one copy of the AGT gene in the mammalian genome, which is ˜11,800 base pairs (bp) in length.1,2 This gene has 5 exons that encode for the protein, separated by 4 introns. Exon 1 codes for the 5′-nontranslated region, whereas exon 2 contains the signal peptide and coding regions. AGT is secreted constitutively from hepatocytes; however, several factors (e.g., glucocorticoids, estrogens, thyroid hormone, insulin, and A-II) may exert a positive feedback.3,4
RELATIONSHIP TO HYPERTENSION
A high molecular-weight form of AGT, which is released during pregnancy, may play a role in pregnancy-induced hypertension. In adipose tissue, there is a form of AGT that is increased by insulin and decreased by 3-adrenergic blockade and that possibly contributes to obesity-related hypertension. Moreover, AGT may play a role in certain other forms of hypertension, as observed in glucocorticoid excess states (e.g., Cushing syndrome) and thyroid disorders.1,2,3 and 4 Interestingly, antisense nucleotide sequences that have been used to block AGT mRNA can reduce blood pressure. In spontaneously hypertensive rats, central nervous system (CNS) administration of antisense sequences against the mRNA encoding AGT lowers blood pressure.5 Furthermore, rats made transgenic with human AGT develop hypertension because AGT is expressed in the blood vessel wall.6,7 Thus, there is ample documentation for a role of AGT in experimental hypertension.
Further documentation for a role of AGT in hypertension is as follows: Epidemiologic studies have shown a relationship between plasma AGT and human hypertension.8,9 Polymorphisms of the AGT gene have been linked to familial hypertension, renal disease, and cardiovascular risk factors.9 Increased levels of AGT are associated with essential hypertension, and an M235T polymorphism in the AGT gene is associated with nephropathy in type 2 diabetics.10 The AGT M235T molecular variant—threonine substituted for methionine at amino acid 235—is associated by linkage analysis with essential hypertension, especially in whites.11,12 Subjects bearing the 235 allele have higher levels of AGT (i.e., a 20% increase in homozygous subjects [TT] and a 10% increase in heterozygous subjects [MT]) compared with homozygous (MM) individuals. This linkage of AGT variants to hypertension is population-dependent (i.e., strong in whites, weak in Mexican Americans13 and the Chinese14). AGT mutations probably play a significant but modest role in blood pressure variation. All of these findings suggest that the renin-AGT reaction kinetics are increased by AGT variants, leading to more A-II production, which in turn may increase vascular resistance and growth. In turn, vascular injury induces AGT gene expression in the vascular media and neointima, suggesting that the renin-angiotensin system participates in myointimal proliferation.15 Finally, gene knock-out mice for AGT develop hypotension with polyuria when challenged with a high-salt diet.16
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


