Forces
Strength
Range (nm)*
Specificity
Main factors
vdw forces
Weak
0–10
No
Interface complementarity
H-bond
Moderate
<0
Partial
Hydrogen donor/acceptor at interface
Electrostatic forces
Moderate
0–10
No
Charge state, ionic strength
Hydrophobic interaction
Strong
0–10
Partial
Hydrophobic surface
π–π stacking
Strong
0–5
Yes
Aromatic ring orientation
Salt bridge
Strong
<1
Yes
Multiple recognition
Clearly, it is important to develop a fundamental understanding of the thermodynamic parameters behind NP-protein interactions. Isothermal titration calorimetry (ITC) is a powerful tool for investigating the fundamental thermodynamics at the NP-protein interface. A model based on ITC results [11] describes the overall complexation process between NPs and proteins using the following equations:
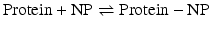
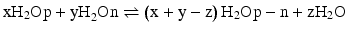

(1)
(2)
(3)
Here, H2Op, H2On, and H2Op-n denote water molecules associated with the protein, NP, and NP-protein complex, respectively. Equation 1 describes NP-protein complex formation, where the changes in enthalpy (ΔH) and entropy (ΔS) are negative. Equation 2 refers to the solvent reorganization process involved in NP-protein complexation, where ΔH > 0 and ΔS > 0. Depending on the contribution of these two processes, the overall complexation (Eq. 3) of NP and protein can be either endothermic (ΔH > 0 and ΔS > 0) or exothermic (ΔH < 0 and ΔS < 0). Therefore, these results suggest that for NP-protein complexation, enthalpy and entropy can be balanced to get a favorable free energy change (ΔG < 0). In the following sections, we will discuss how NP-protein interactions can be tuned by changing the chemical functionalities at the NP surface and how this affects the structure and function of the proteins.
2.1 Reversible versus Irreversible Interactions
Proteins can either bind to NPs reversibly or irreversibly. While both modes of interaction can alter protein structure and behavior, irreversible binding presents by far a greater challenge in understanding and in functional applications of nanomaterials. In order to be functional, proteins must retain their structures and activities. The denaturation of proteins on particle surfaces gives rise to a “hard” protein corona that can dramatically alter particle behavior in vivo.
In early studies, anionic NPs were used to inhibit the enzymatic activity of proteins, such as α-chymotrypsin (ChT) [12]. These NPs interacted electrostatically through cationic patches at the active site of this protein. Enzymatic inhibition was followed by slow irreversible denaturation of the secondary structure of the protein. The structural denaturation of this protein was due to the interaction of the nonpolar interior of the NP with the hydrophobic residues of ChT. Most applications of protein-NP conjugates require retention of protein activity, making retention of structure an important priority. The surfaces of NPs can be appropriately tailored to prevent the structural denaturation of proteins. For instance, introduction of oligo(ethylene glycol) (OEG) [13] functionality on the NP surface drastically reduces the rate of denaturation of ChT.
An interesting area of research in bioconjugate chemistry is the generation of ensembles, where the collective properties of the conjugate structure differ from the properties of the individual precursor components. Enzymatic inhibition of ChT by anionic NPs arises not only from the blocking of the ChT active site by the NPs, but also depends on the charge state of the substrate (Fig. 1). The interaction of ChT with anionic NPs leads to the three-fold increase [12] in the catalytic activity of ChT for cationic substrates, while reducing its activity by 50 and 95 % for neutral and anionic substrates, respectively. This phenomenon of substrate selectivity can be attributed to the unique electrostatic environment presented by the protein-NP conjugate.
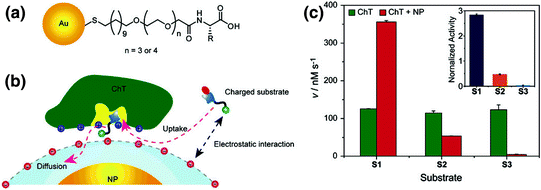
Fig. 1
a The chemical structure of anionic amino acid-functionalized gold nanoparticles. b Schematic representation of the interaction between negatively charged NPs and ChT, illustrating the effect of the charge not only on the binding, but also on the regulation of the enzymatic activity. c Generation rates for each substrate (S1 cationic, S2 neutral, and S3 anionic) as evidence that the binding selectivity is due to electrostatic interactions (Reproduced with permission from reference [12] © 2006 American Chemical Society)
2.2 Structural Implications of Protein-Nanoparticle Interactions. Nanoparticles Can Stabilize or Destabilize Bound Proteins
The stability of NP-ChT complexes has been studied in greater detail using amino acid-functionalized gold nanoparticles (AuNPs). The functionalization of amino acids on AuNP surfaces provides direct access to biomimetic structural diversity. To probe the interaction between ChT and amino acid functionalized AuNPs, ChT catalyzed hydrolysis of N-succinyl-L-phenylalanine p-nitroanilide (SPNA) [14] was first examined in the presence of different concentrations of NPs. All anionic amino acid-functionalized AuNPs showed enzymatic inhibition (Fig. 2a), indicating that complementary electrostatic interactions are essential for ChT-AuNP complex formation. AuNPs with polar side chains, such as aspartic acid (Asp), glutamic acid (Glu), and asparagine (Asn), inhibited ChT activity by ~80 %, while NPs with hydrophobic side chains inhibited ChT activity by ~60 %. Presumably, the hydrophobic active site of ChT was more accessible to SPNA when the hydrophobic amino acids were present on the AuNP surface in comparison to when it was surrounded by NPs bearing hydrophilic side chains. The binding constants of the ChT-NP complexes were also dependent on the surface functionalization of the NPs. NPs with hydrophilic side chains, such as Asp-NP, Glu-NP, Asn-NP, and Gln-NP, showed a binding constant of 3 × 106 M−1, whereas Met-NP had a binding constant of 1.3 × 107 M−1, proving that hydrophobic interactions assisted in ChT-NP complex formation.
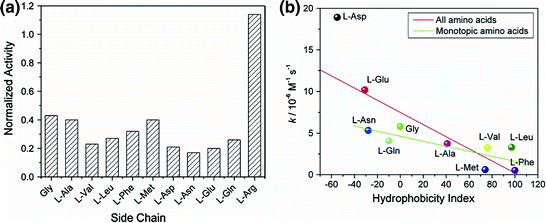
Fig. 2
a Normalized activity of ChT (3.2 µM) with nanoparticles (0.8 µM) bearing various amino acid side chains. b Correlation between the denaturation rate constants of ChT and the hydrophobicity index of amino acid side chains in nanoparticles (Reproduced with permission from reference [14] © 2006 American Chemical Society)
Another important concern that needs to be addressed is the influence of amino acid-functionalized NPs on protein conformation. Circular dichroism (CD) and fluorescence studies showed that NPs with hydrophobic side chains such as Phe-NP, Leu-NP, Met-NP, Val-NP, and Ala-NP, had very little effect on the native structure of ChT. On the other hand, NPs with hydrophilic side chains such as Gln-NP, Asn-NP, Asp-NP, and Glu-NP, showed drastic change in the conformation of ChT. As expected, Arg-NP had no effect on the native structure of ChT. These observations strongly suggest that the carboxylate functional groups facilitated the denaturation process. Competitive H-bonding interactions might destabilize the α-helical structure of ChT leading to the disruption of its secondary structure. Moreover, the above observation could be attributed to the fact that the hydroxide ions in the dianionic side chains were involved in the breakage of the salt bridge between the N-terminus of Ile16 and the Asp194 side chain. A plot of the denaturation rate constants of ChT with various NPs versus the hydrophobicity index of amino acid side chains (Fig. 2b) showed that the dianionic side chains strongly increased the rate of denaturation. The other side chains showed lesser, but still evident, correlation between hydrophobicity and denaturation rate, indicating that the hydrophobicity of the amino acid side chains on the NPs played a role in ChT denaturation/stabilization.
On the contrary, functionalized NPs can also act as chaperones to refold denatured proteins. Rotello et al. have used highly charged 2-(10-mercaptodecyl)malonic acid-functionalized AuNPs (AuDA) (Fig. 3a) [15] to fold thermally denatured cationic proteins, such as ChT, papain and lysozyme. Thermal denaturation exposes the hydrophobic internal core of these proteins. Upon application of AuDAs, NP-protein complexes are formed via electrostatic interactions between the NP and the positive residues of the protein. The high negative charge of the AuDA prevents aggregation and thus facilitates refolding. After partial folding, proteins can be released from the NPs by increasing the ionic strength of the solution; they then self-fold into their native structures (Fig. 3b).
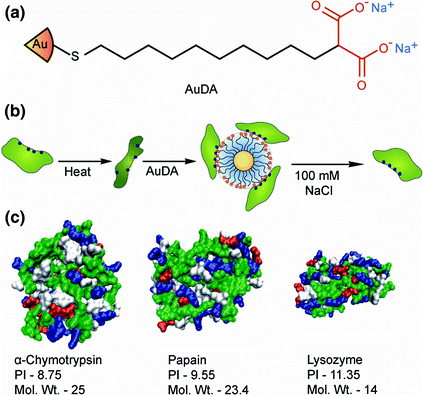
Fig. 3
a Schematic representation of the structure of AuDA and b the NP-mediated refolding of proteins after thermal denaturation. c Space filling models showing the surface structures of the three positively charged proteins used in the refolding study (Reproduced with permission from reference [15] © 2008 The Royal Society of Chemistry)
3 Delivery of Functional Proteins
Cells contain thousands of important proteins that participate in cellular functions [16]. The malfunctioning proteins can induce diseases [17]; thus, protein therapy is emerging as a promising approach to treat these illnesses. The success of this approach is, however, hindered by biological barriers, including inefficient delivery, endosomal entrapment, and proteolysis of proteins. One well-recognized strategy to overcome these challenges is through the use of nanocarrier-based drug/biomolecule delivery systems. Nanocarriers can protect the drug/biomolecules against degradation, control the pharmacokinetics/biodistribution of proteins for maximizing potency while minimizing attributed side effects, and enhance plasma half-life, reducing the frequency of drug administration to enable higher patient compliance [18]. An efficient nanocarrier for anticancer use has to pass several barriers, including the clearance by the reticuloendothelial system (RES) [19] and the crossing of the peritumoral endothelium [20] and the dense extracellular matrix (ECM) that often exists in solid tumors [21]. A primary driving force in nanoscale drug delivery is the enhanced permeability and retention (EPR) effect that relies on the porous architecture and poor lymphatic drainage of tumor vasculature [22]. Several years of research on nanoparticulate drug delivery systems have helped scientists to ascertain several design criteria that can be used to improve the functionality of NPs in vivo. Smaller NPs (>100 nm) are less likely to be engulfed by RES cells. Furthermore, hydrophilic polymers, such as poly(ethylene glycol) (PEG), when grafted to the surface of NPs, will increase circulation times leading to higher targeting capabilities of nanocarriers. To further increase the targeting capability of NPs, vector molecules (e.g., antibodies, aptamers, and small ligands) can be attached to their surface. It is noteworthy that tumors are composed of a heterogeneous population of multiple cancer cells [23]. Thus, multifunctional nanotherapeutics should be designed to target multiple cell lines in parallel.
Recent advances in peptide/protein drug development in cancer research include the introduction of antigens as cancer vaccines (e.g., PROVENGE is the first FDA-approved cancer vaccine for the prevention of prostate cancer) [24] and the application of cytokines, such as interferons (IFN)-β1a, IFN-α2a, granulocyte colony stimulating factor (GCSF), tumor necrosis factor(TNF)-α, and interleukins (ILs), along with small molecule drugs for the clinical treatment of cancer. Most importantly, monoclonal antibodies kill cancer cells by antibody-dependent, cell-mediated cytotoxicity and many are under consideration in clinical trials for the targeted therapy of breast, lung, colorectal, gastric, and brain solid tumors, metastatic events, and hematological malignancies. Indeed, several antibodies have already found their way into the clinic. Obviously, devising ways to specifically deliver such molecules to certain targets within the human body is an emerging area of research in NP-mediated drug delivery.
3.1 In Vitro
Specific nanomaterials, such as liposomes, polymeric systems, and even biomimetic particles have been proposed for the intracellular delivery of proteins for cancer therapeutics. However, the delivery of proteins using these vectors can be limited by instability and alteration of protein activity.
3.1.1 Inorganic NPs
Nanoparticles (NPs) have been widely considered for protein delivery applications. For instance, Ghosh et al. [25] have prepared gold NPs coated with short peptides, which effectively noncovalently complexed and delivered active anionic proteins (e.g., β-galactosidase) into a variety of cell lines (COS-1 (monkey kidney cells), MCF7 (human breast cancer cells), and even hard-to-transfect muscle cells (C2C12)). The engineered gold NPs were composed of interior alkyl chains for promoting core stability, a corona of tetraethylene glycol (TEG) for the prevention of denaturation and nonspecific interactions with biomolecules, and external arginine-rich peptide-tags working as a recognition unit. The authors hypothesized that the release of β-galactosidase from the particles was mediated by glutathione, which is present in the intracellular environment.
3.1.2 Nanocapsules
Hollow nanocarriers (e.g., nanocapsules and nanoliposomes) can provide more efficient systems for protein delivery applications due to their enhanced payload capacity. Besides protection against degradation by pH and light, hollow nanocarriers can also help reduce tissue irritation and provide more controllable targeting release of therapeutic agents. Tang et al. [7] fabricated nanocapsules for the delivery of functional proteins and enzymes to the cytoplasm. The functional proteins were incorporated in a capsule shell. The effectiveness of these capsules in the cytoplasmic delivery of proteins was confirmed by the delivery of fully functional caspase-3 to HeLa cells (Fig. 4) with concomitant apoptosis.
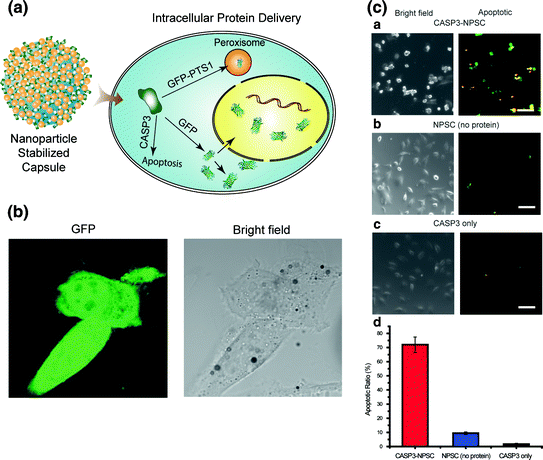
Fig. 4
A A schematic representation of the intracellular delivery of GFP and caspase-3 (CASP3) using nanoparticle-stabilized capsules (NPSCs). B Confocal images showing GFP delivery into HeLa cells by NPSCs (Scale bars: 20 μm). C Delivery of caspase-3 into HeLa cells. Cells were incubated for 1 h with (a) CASP3-NPSC, (b) NPSC without CASP3, and (c) only CASP3 without NPSC. Subsequently, cells were stained using Yopro-1 (green fluorescence) and 7-AAD (red fluorescence) for 30 min, and the overlapped images are presented as apoptotic. (d) Apoptosis ratios of the cells after CASP3 delivery (Scale bars: 100 μm). The error bars represent the standard deviations of three parallel measurements (Reproduced with permission from reference [7] © 2013 American Chemical Society)
In another study, Yan et al. [26] produced nanocapsules composed of a protein core (e.g., green fluorescent protein (EGFP), horseradish peroxidase (HRP), bovine serum albumin (BSA), superoxide dismutase (SOD) and caspase-3), and a thin permeable polymeric shell anchored covalently to the protein core. The pH sensitive shell of the nanocapsules enabled controlled delivery of the proteins at specific pH values. The size, surface charge, and degradability of the nanocapsules could be tuned by varying the composition of the core, the nature of the crosslinkers as well as the type of employed monomers in construction of the shell. While non-degradable capsules showed long-term stability, the degradable ones released their ingredients inside the cells after breaking their shells. This system was shown to deliver multiple proteins to cells in a nontoxic and highly efficient manner, mainly through the caveolae-mediated endocytosis pathway. The cell transduction efficiency of nanocapsules containing EGFP in HeLa was 2–3 times higher than that of TAT–EGFP fusion proteins or antennapedia–EGFP conjugates. Thus, depending on the type of employed core proteins, nanocapsules may be suitable for a variety of biomedical applications, such as cellular imaging, cancer therapies, and anti-aging solutions. For example, the combination of indole-3-acetic acid (IAA) and HRP was recently proposed as a potential prodrug for the treatment of cancer [27]. This prodrug works via the HRP-mediated conversion of IAA into a free radical intermediate, which finally results in the induction of apoptosis in mammalian cells [28]. Only nanoencapsulated HRP, and not the native HRP, could decrease cell viability with increasing IAA concentration.
Yeh et al. [29] synthesized multifunctional NP-stabilized capsules consisting of fluorescent polyhistidine-tagged proteins attached to CdSe/ZnS core–shell quantum dots through metal-affinity coordination. This negatively charged complex could interact with cationic gold NPs, which are anchored to the fatty acid core through guanidinium–carboxylate interactions. This lipophilic core functions as a reservoir for endosome-disrupting agents. The self-assembled system features stimuli-responsive delivery of multiple proteins into cell cytosol combined with FRET-based fluorescence tracking of cytosolic and vesicular distribution (see Fig. 5).
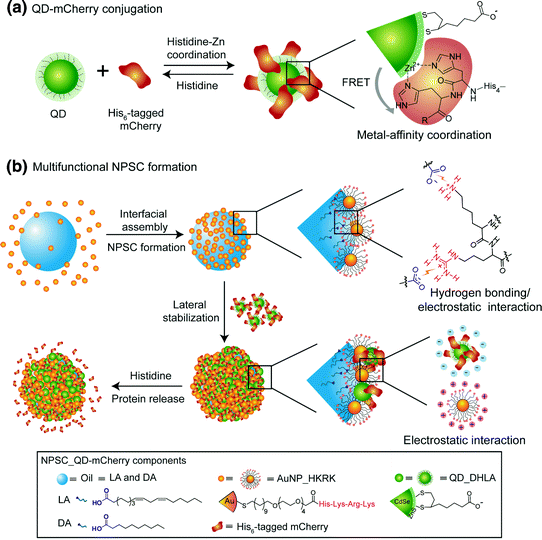
Fig. 5
a Dihydrolipoic acid-functionalized CdSe/ZnS core–shell quantum dots (QD_DHLA) and hexahistidine-tagged (His6-tagged) mCherry were conjugated by the reserved metal-affinity coordination using histidine as competitive molecules. b Fabrication of the template droplets by agitation of AuNP_HKRK (AuNPs functionalized with peptide (histidine-lysine-arginine-lysine)) and oil (linoleic acid (LA) and decanoic acid (DA)) in 5 mM phosphate buffer (pH 7.4). Then, the prepared template droplets were introduced into an AuNP_HKRK solution and crosslinked by anionic QD–mCherry conjugates to form NPSC_QD–mCherry (Reproduced with permission from reference [29] © 2014 Wiley-VCH Verlag GmbH & Co.)
3.1.3 Liposomes
Liposomes, another class of hollow nanocarriers, are vesicle-like structures that can retain therapeutics either in their hydrophilic core or their hydrophobic lipidic layers [30]. In the context of cancer (besides the well-known small molecule Doxil liposomal formulation), liposomes have been widely employed for delivery of various biomolecules, including IL-1, IL-2, IL-6, granulocyte-moncyte colony stimulating factor (GM-CSF), INF-γ [31], and granulocyte colony stimulating factor (GCSF) [32]. Liposomal encapsulation increases the protein half-life and enables the sustained release of encapsulants. For instance, insulin and peptides, such as leuprolide, enkephalin, and octreotide encapsulated in DepoFoam multivesicular liposomes were released at a controlled rate over a period from a few days to several weeks [33]. Liguori et al. [34] formulated pro-apoptotic membrane proteins (containing the voltage-dependent anion channel (VDAC) and pro-apoptotic Bak) into natural proteoliposomes to examine growth inhibitory effects in human colorectal carcinoma cells. The recombinant proteins were integrated into the lipidic bilayer of the liposomes. The induction of apoptosis was detected after a 24 h incubation period, which was accompanied by cytochrome c release and activation of caspases-3, -7, and -9 together with poly ADP ribose polymerase (PARP). The VDAC regulates the release of cytochrome c from mitochondria and Bak controls the permeability of mitochondrial membrane upon activation. The uptake of recombinant proteoliposomes by mammalian cells induces apoptosis by release of cytochrome c and activation of caspases. Gao et al. [35] formulated Pseudomonas exotoxin-based immunotoxins (PE38KDEL) into PEGylated anti-HER2 Fab´-functionalized liposomes, which showed promising results in the treatment of HER2-overexpressing breast cancers in vitro.
3.1.4 Polymeric Nanocarriers
Polymeric systems also have some applications in the field of drug delivery to cancer. 250 nm poly(γ-glutamic acid)-based NPs were used as protein carriers to deliver tumor vaccines, antigenic proteins, to antigen-presenting cells [36]. The ultimate goal of immunotherapy is to fight cancer through amplification of tumor immunity. With such a strategy, the immune system can distinguish the cells which express the tumor-associated antigens (TAAs) from normal cells. However, in this regard, TAA-specific cytotoxic T lymphocytes must be stimulated by the efficient delivery of TAA to antigen-presenting cells. Since antigen presenting cells only incorporate particles with a size range of 50–3 μm [37], nanoparticles can be useful for delivery of TAAs. The poly(γ-glutamic acid) NPs fabricated in this study had higher protein entrapment efficiency of 55–60 %. The NPs were shown to effectively deliver the encapsulated antigenic proteins inside the dendritic cells through cytosolic translocation from the endosomes and the ovalbumin (OVA) antigen was processed into epitope peptides and was subsequently presented via MHC molecules [36]. The authors investigated the inhibitory effects of NP/OVA in the lung metastasis of B16-OVA tumors expressing OVA as a TAA. Triplicate immunization with this formulation decreased the number of lung metastasis nodules in mice even more than Freund’s complete adjuvant/OVA vaccine. Since poly(γ-glutamic acid) NPs are relatively stable in vivo, they could be used as controlled release systems of entrapped proteins. Authors also further analyzed the effect of entrapment process on the activity of catalase as a model protein. It was shown that entrapped catalase retains more than 90 % of its enzymatic activity [36].
3.1.5 Biomimetic Particles
Virus-like particles (VLPs) provide biogenic vectors for delivery. Kaczmarczyka et al. [38] used VLPs derived from an avian retrovirus for delivery of proteins to different target cells. The VLPs can be used to deliver proteins either as part of Gag fusion proteins (for intracellular delivery) or on the surface of VLPs. Since avian retroviruses do not replicate in human cells, the safety of this system for use in human applications is potentially high. VLPs containing Gag-Cre recombinase, Gag-Fcy:Fur, and Gag-human caspase-8 were synthesized. The authors successfully delivered caspase-8 to PC3 cells. Furthermore, in addition to the cellular delivery of proteins, in the same study, it was shown that murine IFN-γ and human TNF-related, apoptosis-inducing ligand (TRAIL) could be displayed on the surface of VLPs, and subsequently, these modified VLPs could activate the appropriate cellular receptors on the surface of cell membranes (in a mouse macrophage cell line developed from a C57BL/6 mouse that expresses IFN-γ receptor and PC3 cells, respectively).
3.2 In Vivo
Many proteins are believed to be promising candidates for cancer therapy; however, their clinical application is hampered by their removal through RES organs (spleen, lymph nodes, and liver) [19], their degradation by proteases [39], and their distribution and accumulation in non-target organs [40]. Furthermore, since high concentrations of particular proteins must be used due to the non-targeted nature of the therapy, the incidence of systemic side effects could be high. Nanoparticulate drug delivery systems could thus reduce the concentration of the drug required for clinical efficacy.
3.2.1 Inorganic NPs
TNF-α [41] was shown to cause hemorrhagic necrosis of solid tumors. Such a surprising finding led scientists to rapidly develop TNF-α for clinical testing; however, studies were discontinued due to some life-threatening toxicity observed in patients. Similar side effects were also later reported for other cytokines. Further attempts to localize the delivery of TNF to solid tumors, by surgical procedures, which could significantly improve its therapeutic index [42, 43], showed that by confining the therapy to the intended target, one may actually increase the efficacy and reduce the side effects associated with this therapy.
In later studies, Paciotti et al. [44], developed 26 nm colloidal gold NPs, on which CYT-6091, a multivalent drug, was mounted and meant to deliver TNF to its target. In this system, TNF was bound to PEGylated gold NPs. TNF not only has antitumor activity per se, but also functions as a targeting moiety and directs the NPs to MC-38 solid tumors (seven–ten-fold increase in targeting efficiency). A mere 7.5 mg of this formulation was shown to be as effective as 15 mg of native TNF. Furthermore, in mice receiving native TNF, the mortality rates were 33 and 15 % (with 15 and 7.5 mg, respectively); none of the animals receiving CYT-6091 died.
To enhance the antitumor efficacy of CYT-6091, which is a single agent therapy, the same authors later designed the CYT-21001 vector, which delivers TNF and paclitaxel simultaneously to the solid tumors [45]. The results of a phase I clinical trial of CYT-6091, consisting of TNF-α covalently linked to PEGylated colloidal gold NPs were published in 2009 [46]. The intravenously (IV) injected formulation has proven to be safe in 29 patients with solid tumors, and it showed a prolonged half-life compared to the native TNF-α and trafficking of the attached protein to the tumor, as assessed by electron microscopy in tumor biopsies.
Brinas et al. [47] have recently produced 3–5 nm gold NPs that were subsequently coated with two tumor-associated glycopeptide antigens (mucin (MUC4) and the Thomsen-Friedenreich antigen). The third coating agent was composed of a 28-residue peptide from the complement-derived protein C3d which functioned as a B-cell activating adjuvant. The immunization of mice with this vaccine led to the production of both IgM and IgG in the serum, showing the potential of this system in designing vaccines using TAAs.
3.2.2 Liposomes
Liposomes show high promise for in vivo applications. For instance, liposomes have been used for the cytosolic delivery of exogenous soluble protein antigens into antigen-presenting cells [48]. In this method, which adopts a cytosol-invading listerial for endosomal escape, listeriolysin O (which is a pore-forming protein that contributes to Listeria monocytogenes escape from the endosome into the cytosol) is co-encapsulated inside liposomes with OVA. Immunization of mice with this construct stimulated the activity of OVA-specific cytotoxic T lymphocytes and increased antigenic peptide-specific cytotoxic T lymphocyte precursor frequency. Overall, vaccination conferred protection to mice from lethal challenges with antigen-expressing tumor cells. The same group later demonstrated killing of B16 melanoma cells by listeriolysin O-liposome-mediated delivery of the protein toxin, gelonin. Gelonin is capable of enzymatically inactivating ribosomes and arresting protein synthesis in cancerous cells [49]. While the liposomal formulation had an IC50 of 0.1 nM with an incubation time of only 1 h, the free gelonin or listeriolysin-free formulation led to no detectable cytotoxicity. Direct intratumoral injection into mice bearing subcutaneous (SC) solid B16 melanoma showed that the listeriolysin O-liposomes were more effective than control formulations in restraining tumor growth.
Cruz et al. [50, 51] showed the enhanced survival of animals bearing PI534 tumors upon treatment with asparginase-encapsulated liposomes. Furthermore, a reduced occurrence of neutralizing antibodies was noted. Further studies by the same group demonstrated the superior efficacy of liposomal IL-2 over free IL-2 in inhibition of experimental M5076 metastases in mice [52]. In another study, liposomes harboring unmethylated cytosine-phosphorothioate-guanine containing oligodeoxynucleotides co-encapsulated with OVA were shown to inhibit the tumor growth and to completely cure ~50 % of OVA-expressing EG-7 tumor-bearing C57BL/6 mice upon intradermal inoculation [53].
A preclinical study demonstrated that VacciMax® (a vaccine composed of liposomes encapsulating human papilloma virus (HPV) 16 E7-derived cytotoxic T lymphocyte epitope fused to the T helper epitope PADRE and combined with CpG or lipopeptide adjuvant) could protect C57BL/6 mice against post-tumor challenge with HPV 16-expressing C3 tumor cells [54]. Neelapu et al. [55] incorporated a TAA, the idiotype of the Ig on B-cell malignancy in dimyristoylphosphatidylcholine lipid liposomes containing IL-2 as an adjuvant and the vaccine was shown to protect mice against lymphoma in preclinical studies. Later, the same group tested the safety and immunogenicity of the vaccine in 10 patients with advanced-stage follicular lymphoma [56]. The liposomal cancer vaccine proved to be safe and induced, sustained antitumor immune response through tumor-specific CD4+ and CD8+ T-cells in patients. After 50 months follow-up, 6 of the 10 patients were reported to remain in continuous first complete remission.
Broekhoven et al. [54] constructed B16-OVA-derived plasma membrane vesicles containing a new metal-chelating lipid, 3(nitrilotriacetic acid)-ditetradecylamine, which was used to engraft the recombinant single chain antibody fragments (ScFvs) to the dendritic cell surface molecules CD11c and DEC-205 onto the vesicle surface. The formulation was shown to stimulate strong B16-OVA-specific cytotoxic T lymphocyte responses in splenic T cells and to protect syngeneic mice against tumor growth.
3.2.3 Polymeric Nanocarriers
Polymeric systems are among the most extensively used platforms for protein therapy. For example, RGD peptide-loaded glycol chitosan NPs modified with 5β-cholanic acid were shown to possess antiangiogenic and antitumoral efficacy against solid tumors in mice upon IV injection or intratumoral administration [57].
Lim et al. [58] employed a formulation containing PEGylated heparin, TRAIL, and poly-L-lysine to increase its short biological half-life, improve its inherent instability, and eliminate its potential hepatotoxicity. IV injection of NPs in HCT-116 tumor-bearing BALB/c athymic mice efficiently suppressed tumor growth (>70 %) and induced significant tumor cell apoptosis without inducing liver toxicity.
PE38KDEL or fusion protein truncated Pseudomonas Exotoxin A has been used to prepare immunotoxin with monoclonal antibodies. Gao et al. [59] fabricated PE38KDEL-I-loaded poly(lactic-co-glycolic acid) (PLGA) NPs conjugated with F(ab’) fragments of a humanized SM5-1 monoclonal antibody (PE-NP-S). After binding to SM5-1 binding protein-expressing hepatocellular carcinoma cell lines, PE-NP-S was internalized by these cells, and induced significant cytotoxicity. Further, in vivo studies on SM5-1 binding protein-overexpressing tumor xenograft model demonstrated that administration of the formulation significantly suppressed tumor development and even induced tumor regression. Due to the increased cancer therapeutic efficacy compared to older counterparts of the drug as well as reduced nonspecific toxicity and immunogenicity, this formulation might be a promising modality for cancer therapy.
Other studies have also revealed that poly(methoxypolyethyleneglycol cyanoacrylate–co-n-hexadecylcyanoacrylate) NPs could enhance the half-life of TNF-α in tumor bearing mice along with the targeting efficiency and antitumor potency [60]. The increased antitumor activity might arise from the higher accumulation of nanotherapeutics in tumor tissues and longer plasma circulation time.
4 The Protein Corona
As discussed in the introduction, protein-particle interactions that generate the protein corona are an important general issue in therapeutic delivery. Plasma proteins play crucial roles in blood, and they are found at high concentrations in the bloodstream. The layer of proteins that instantaneously covers NP surfaces when NPs enter biological fluids (e.g., blood, plasma, cell culture media, and intracellular environment) is called the protein corona. Even NPs functionalized with vector proteins (not considered as a corona) would pick up an additional layer of corona proteins upon entrance to a biological fluid. Protein coronas have the capability to significantly affect the biokinetics and in vivo fate of NPs. The adsorption of proteins lends a new bio-identity to NP surfaces, influencing their biological interactions. NP size [61], NP shape [62], and several surface characteristics, including the presence of surface functional groups [63], surface topography, uniformity, roughness [64], and surface hydrophobicity/hydrophilicity, [65–67] influence the nature of proteins adsorbed in the protein corona. Furthermore, interaction temperature [63, 68], NP dispersal environment [69], protein source [70], incubation time with protein source [71], concentration of protein source (proteome variability) [71–73], and gradient concentration [74] can also influence the corona composition. In turn, depending on the nature and amount of specific proteins adsorbed, NP biokinetics and their fate will be different. The adsorption of proteins to the surfaces mainly stems from the increase in the total entropy of the proteins on the surface as well as the nonspecific interactions between the proteins and NP surface [75]. While proteins with isoelectric points (pI) < 5.5 primarily bind to NPs with basic surfaces, those with pI > 5.5 are bound to NPs with acidic surfaces [76]. However, it is noteworthy that the adsorption of proteins to some surfaces might also be specific. For example, in the case of spherical nucleic acids, different sequences lead to formation of coronas with different compositions and this subsequently affects the level of cellular uptake [77]. The characterization of corona composition and its evolution in biological systems has been challenging due to the enormous complexity of the proteins and their interactions on NP surfaces [78].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


