The Immune System and Immunotherapy
21.1 INTRODUCTION TO THE IMMUNE SYSTEM
One feature that is common to all organisms is the ability to defend themselves against challenges in the environment in which they live. Mammals have a complex phalanx of defenses against bacteria, viruses, and parasites, which comprise the immune system. The immune system can be very broadly characterized as having 2 major arms: innate immunity and adaptive immunity. The innate immune system is the “first line of defense” against pathogens, and includes macrophages and dendritic cells that function in part to present antigens to the cells in the adaptive immune system. The adaptive arm of the immune system is mediated by lymphocytes and responds with higher specificity to pathogens. Key cells of the adaptive immune system are helper T cells (Th) that express a marker known as CD4, cytotoxic T lymphocytes (CTL) that are distinguished by the CD8 marker, and B cells that produce antibodies. The molecular components of pathogens that are recognized by T and B cells are broadly referred to as antigens.
The adaptive immune system has evolved to respond to an initial encounter with a variety of foreign pathogens as well as a potential secondary encounter with the same pathogen. These challenges have shaped the development of the immune system to have the properties of diversity, specificity, and memory. Diversity enables an individual to respond to a broad array of possible pathogens, while at the same time generating exquisite specificity to elements of specific pathogens, ensuring a focused response to a given pathogen while minimizing collateral damage to the host tissues. Memory is the ability of the immune system to respond rapidly to a pathogen that has previously been encountered, thus avoiding sickness and quickly clearing the offending organism without damaging the host.
Another feature of the immune system is that it generally does not attack the host’s own tissues. This recognition of “self” versus “non-self” and the ability to avoid attacking self-tissues is referred to as self-tolerance. Although these mechanisms of tolerance are critical to avoid autoimmunity, they are impediments that need to be overcome in antitumor immunity.
21.2 INNATE IMMUNITY
21.2.1 Antigen Presentation
One of the main functions of the innate immune system is to present antigens to the adaptive immune system to orchestrate a functional immune response. Dendritic cells (DCs) are highly specialized and efficient antigen-presenting cells (APCs). One key function that differentiates DCs from other “professional” APCs, such as macrophages and B cells, is their ability to activate naïve T cells. Antigens are presented to T cells by major histocompatibility complex (MHC) molecules. The MHC consists of a series of proteins encoded by highly polymorphic codominantly expressed genes. As a result, each individual expresses a particular combination of MHC alleles that is different between individuals, and a huge diversity exists in populations. There are 2 main types of MHC molecules. MHC class I molecules are expressed on almost all cells. MHC class II molecules are expressed primarily on APCs such as macrophages, DCs and B cells, or can be induced to be expressed on cells only in the case of inflammation. The mouse MHC class I molecules are referred to as H-2K, H-2D, and H-2L; the mouse MHC class II molecules are I-A and I-E. The human MHC is comprised of 3 class I molecules, HLA-A, HLA-B, and HLA-C and 3 class II molecules, HLA-DQ, HLA-DP, and HLA-DR. Class I heterodimers comprise a transmembrane α-chain noncovalently bound to the nonpolymorphic β2-microglobulin protein (β2m). The class II MHC molecule is expressed on the cell surface as an α and β heterodimer. A schematic of MHC class I and II molecules is depicted in Figure 21–1.
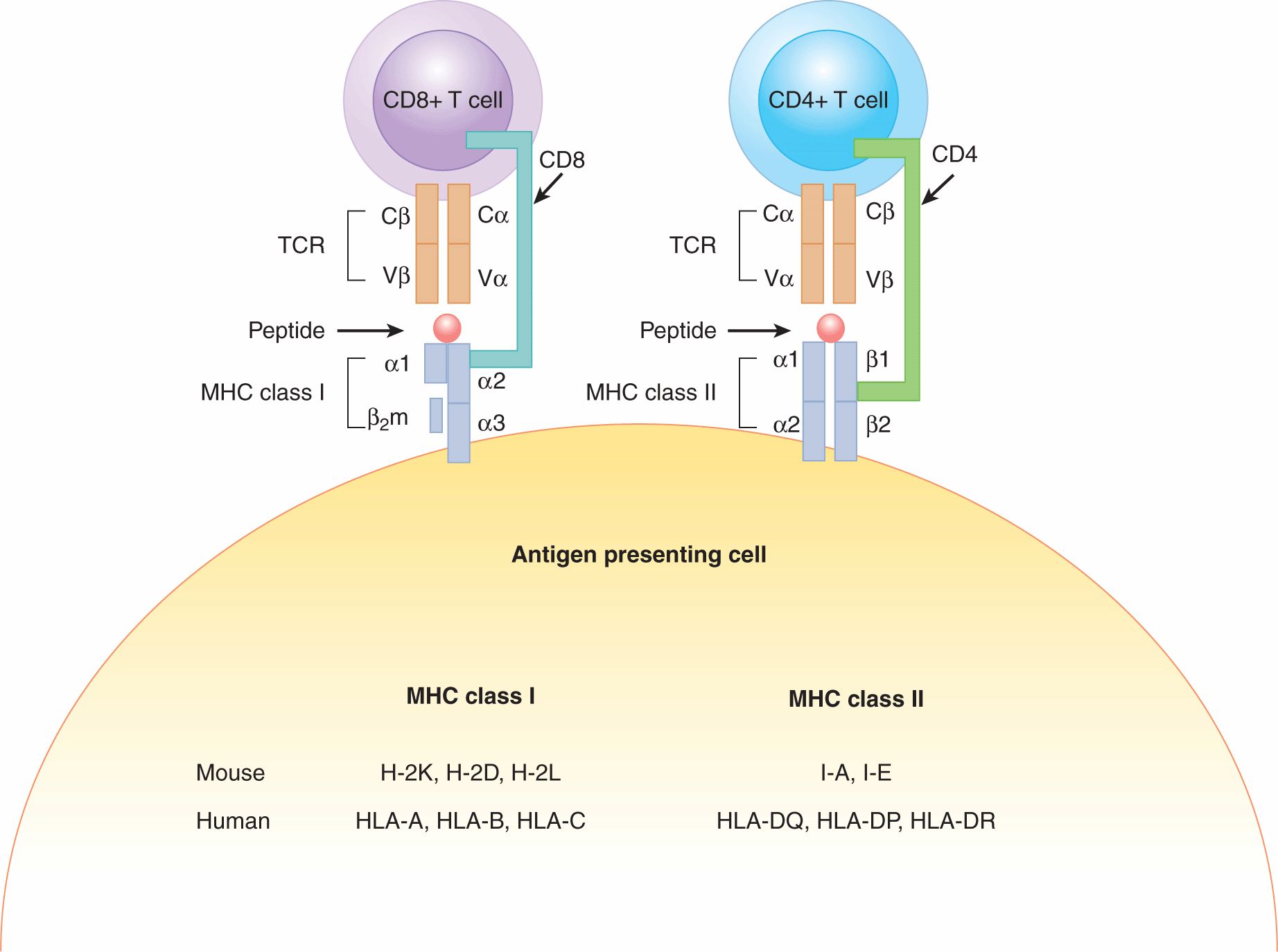
FIGURE 21–1 T-cell recognition of antigens in the context of MHC molecules. The α/β dimer of the T-cell receptor (TCR) recognizes peptide fragments on the surface of antigen-presenting cells (APCs). The TCR on CD4+ T cells binds MHC class II molecules bearing an antigenic peptide. This interaction is stabilized by the CD4 coreceptor from the surface of the CD4+ T-cell binding to the MHC class II molecule. The TCR on CD8+ T cells binds MHC class I molecules bearing an antigenic peptide. This interaction is stabilized by the CD8 coreceptor. The nomenclature of MHC alleles in human and mouse is listed at the bottom of the figure. β2m, β2-microglobulin.
The functions of MHC class I and II molecules are to bind and display peptide fragments for T-cell recognition. These peptides may be derived from self proteins or foreign proteins. The peptide binding cleft of MHC molecules contains the regions of highest polymorphism within the molecule and has an impact on the array of peptides that may be bound. Class I and class II differ in the size of peptides that can bind the cleft. MHC class I binds smaller peptide fragments (8 to 11 amino acids) whereas class II presents larger fragments of proteins (15 to 18 amino acids).
The 3 major pathways by which peptides can be processed and loaded into the binding cleft of MHC molecules are referred to as the exogenous, endogenous, and cross-presentation pathways. These pathways are depicted in Figure 21–2. The exogenous pathway is the predominant pathway for class II loading, whereas the proteins processed through the endogenous and cross-presentation pathways result in class I loading. The endogenous pathway occurs in most cells, whereas the other 2 pathways (exogenous and cross-presentation) occur primarily in APCs.
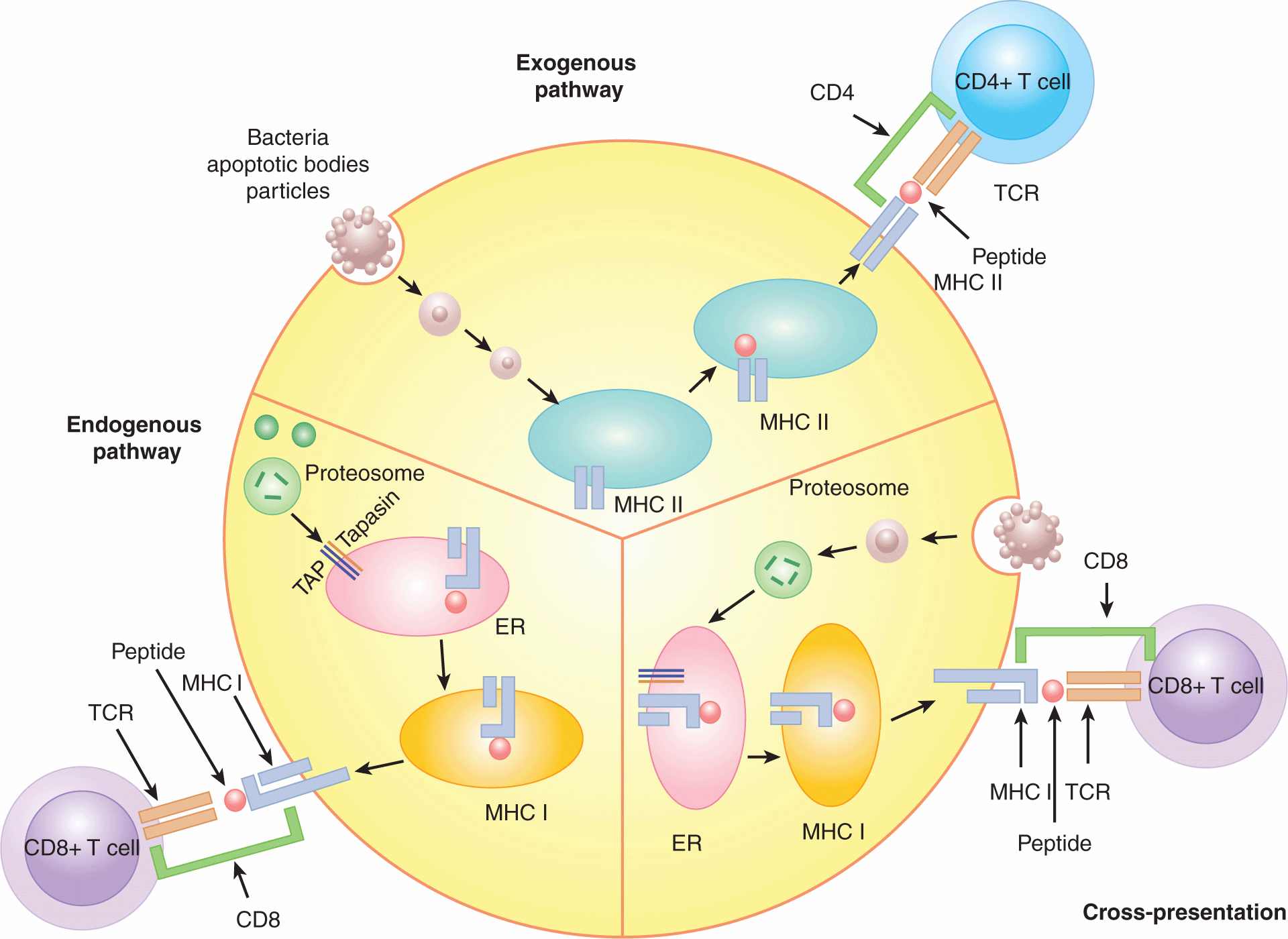
FIGURE 21–2 Antigen-processing pathways in the APC. There are 3 pathways of antigen processing by the APC: exogenous, endogenous, and cross-presentation. The exogenous pathway processes proteins produced outside the APC and places their peptides on MHC class II for recognition by CD4+ T cells. Proteins derived from apoptotic bodies, bacteria, and particulate antigens are processed into peptides by the exogenous pathway. The exogenous pathway occurs in phagocytic cells, including B cells, macrophages, and dendritic cells. The endogenous pathway places cell-produced peptides into the peptide-binding groove of MHC class I for recognition by CD8+T cells. The endogenous pathway is responsible for immune recognition of viral peptides or self peptides. Peptides are cleaved from proteins in the cytosol by the proteasome. They then enter the endoplasmic reticulum (ER) in a process dependent on the transporter associated with antigen processing (TAP) and are loaded onto MHC class I, which is then shuttled to the cell surface. This pathway occurs in most cells, not just APCs, allowing sensing of viral infection in all cell types. The cross-presentation pathway also loads peptides onto MHC class I for recognition by CD8+T cells, but the proteins that are processed are not cell-intrinsic, and are instead taken up from the surrounding environment. This pathway is important for detecting viruses that infect cells other than APCs and tumor antigens taken up in the form of tumor apoptotic bodies. The cross-presentation pathway is most efficient in dendritic cells.
The exogenous pathway of protein processing begins when an APC acquires material from outside the cell by phagocytosis or receptor-mediated endocytosis. This exogenous material is processed initially in an acidified early endosome by pH-sensitive proteases. The endosome containing the fragmented protein finally fuses with a vesicle containing newly formed MHC class II molecules where the peptides then bind in the cleft of the MHC molecule. MHC class II is produced in the endoplasmic reticulum. When the protein is produced a chaperone protein known as the invariant chain initially occupies the peptide-binding groove. As the MHC/invariant complex leaves the Golgi apparatus, the invariant chain targets the complex to enter the endosomal pathway where proteases known as cathepsins digest the invariant chain until only a small fragment known as CLIP (class II-associated invariant chain-derived peptide) remains in the binding cleft. The MHC II-CLIP complex next associates with proteins that aid in the release of CLIP, freeing the peptide-binding groove for occupancy by the extrinsically obtained peptides. After this peptide exchange, the complex is shuttled to the cell membrane where it is available to be recognized by CD4+ Th cells (Bryant and Ploegh, 2004; Trombetta and Mellman, 2005).
The endogenous pathway is responsible for loading cell-intrinsic proteins in the binding cleft of MHC class I molecules. Proteins are digested by a large macromolecular complex known as the proteosome (see Chap. 8, Fig. 8–9 and Chap. 9, Fig. 9–4). The proteosome normally processes self proteins but when a cell is exposed to inflammatory cytokines, such as interferon-γ (IFN-γ) or tumor necrosis factor (TNF)-α the structure of the proteosome is altered into a structure known as the immunoproteosome, which is more efficient at processing peptides with high binding efficiency to the MHC class I peptide-binding cleft. Self proteins are cleaved into short fragments by the proteosome in the cytoplasm and are then shuttled into the endoplasmic reticulum by the TAP protein complex. As the peptides are guided into the ER by the TAP transporter, they are loaded onto empty MHC class I molecules, which are held close to the TAP complex by the protein Tapasin. In the case of viral infections, peptides derived from the virus will be processed and loaded onto the MHC class I molecule resulting in viral detection by CD8+ T cells. In the case of tumor cells, proteins derived from the tumor cell will be processed and loaded onto MHC class I via the cross-presentation pathway (Pamer and Cresswell, 1998).
Efficient activation of T cells depends upon the acquisition of antigens by APCs and the subsequent presentation of selected peptides in combination with MHC molecules on the cell surface for recognition by T cells. Because MHC class I-restricted peptides are derived from proteins produced intrinsically by the cell, a conundrum arises as to how a T-cell response can be initiated against viruses or bacteria that infect cell types other than APCs. A process known as cross-presentation can occur where cell-exogenous proteins can be taken up by DCs and presented on MHC class I molecules (see Fig. 21–2). This pathway is critical to activation of tumor-specific T cells, as the tumor cells are not usually APCs. The presence of measurable T-cell responses to many tumors implies that at one point a DC may have acquired tumor-derived proteins and processed them into MHC class I-restricted peptides, resulting in the activation of naïve tumor-reactive CD8+ T cells (Shen and Rock, 2006).
21.2.2 Maturation of Antigen-Presenting Cells
APCs can exist in an immature state or a mature (ie, activated) state (Fig. 21–3). Maturation of APCs occurs via specialized cell-surface receptors or intracellular receptors that recognize pathogen associated-molecular patterns (PAMPs). These receptor classes include Toll-like receptors (TLRs), nucleotide binding domain and leucine-rich repeat receptors (NLRs), the retinoic acid-inducible gene-like receptors (RLRs), and c-type lectins (Iwasaki and Medzhitov, 2010). The best-studied receptors are the TLR family of proteins (Takeda and Akira, 2005). The ligands for TLRs include viral/bacterial DNA and RNA, bacterial lipids and endogenously derived molecules, such as heat shock proteins and uric acid crystals that alert the immune system to tissue damage. There are at least 11 identified TLRs in mammals, 9 of which are conserved between human and mouse (TLR1 to TLR9). TLR4 is one of the best-characterized TLRs; the pathogen-associated ligand for TLR4 is lipopolysaccharide (LPS), a molecule found on the outer membrane of Gram-negative bacteria. Several TLR stimuli are being investigated for use as immune adjuvants in humans. For example, the drug imiquimod (see Sec. 21.3.3), that is used as a treatment for warts, activates TLR7. Signaling through TLR7 either by drugs such as imiquimod, or from viral infection, results in production of interferonα TLR9 recognizes unmethylated CpG (cytosine phosphate guanine) DNA structures (Hemmi et al, 2000), that are plentiful in bacterial DNA but rare in mammals. Activation of TLR9 by CpG DNA results in production of the cytokines interleukin (IL)-12 and TNFα which contribute to the induction of cellmediated immunity.
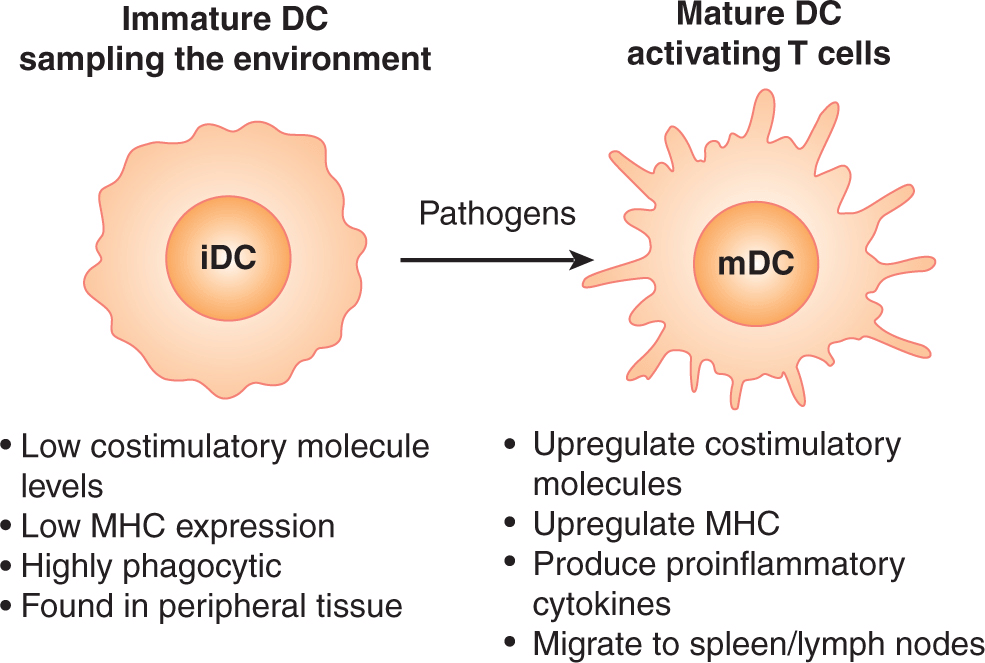
FIGURE 21–3 DC maturation and migration. The DC is the most effective sentinel for the immune system, migrating throughout peripheral tissues seeking damage or infection. The immature DC (iDC) has low expression of costimulatory molecules and MHC. This limits inappropriate activation of T cells in the absence of infection or tissue damage. The iDC is highly phagocytic, sampling the local environment for antigens. Upon encounter with a maturation signal such as ligands for the Toll-like receptor (TLR) family molecules or ligation of the CD40 molecule, the iDC increases levels of costimulatory molecules and MHC and migrates to the local draining lymph node where it can interact with and activate T cells. mDC, Mature dendritic cell.
The NOD-like and RIG-I-like families of proteins (NLRs and RLRs) detect the presence of bacteria and viruses, respectively. NLR proteins sense bacterial products in the cytoplasm by interacting with the C-terminal leucine-rich repeat regions (LRR). Bacterially-derived products bind to the LRR, resulting in a conformational change in intracellular NOD proteins. This change activates the NOD proteins resulting in recruitment of signaling kinases and activation of Caspase (CASP)-1 through binding to caspase recruitment domains (CARDs). This activation results in both de novo cytokine transcription via activation of the nuclear factor-kappa B (NF-κB) and AP1 transcription factors (see Chap. 8, Sec. 8.2.6) and activation of IL-1 through CASP-1 activity (Franchi et al, 2009). RLRs are also localized in the cytoplasm but bind viral-derived RNA sequences. The RLR family contains several members, such as RIG-I, MDA-5, and LGP2. These proteins bind modifications found in the 5′ end of virally produced RNAs that are not found in mammalian RNA. Detection of viral RNA in the cytoplasm results in activation of NF-κB and IRF3 (interferon regulatory factor 3), thereby inducing inflammatory cytokine and type-1 interferon production by the infected cell (Kawai and Akira, 2008).
After the detection of pathogens via these different receptor families, the APC becomes mature and upregulates expression of MHC class II and other costimulatory molecules that contribute to the activation of T cells (see Fig. 21–3). In addition, mature APCs are induced to secrete cytokines and chemokines that promote immune function in a variety of ways. Signaling through TLRs also induces a change in migration of the APC. DCs will change their responsiveness to chemokine signals and migrate out of the peripheral tissues and localize (“home”) to lymph nodes, thus increasing their chances of encountering a T cell with specificity to the pathogen that activated the DC. Monocytes and macrophages will migrate in the opposite pattern, entering the infected or damaged tissue where they take part in pathogen clearance or tissue repair, at the same time acting as local APCs to maintain activation of T cells at the site of infection.
21.3 ADAPTIVE IMMUNITY
21.3.1 Generation of Lymphocyte Diversity
B cells and T cells express highly specific receptors that recognize antigen. The B-cell receptor (BCR) and T-cell receptor (TCR) are generated uniquely in each cell as the consequence of genomic DNA rearrangements. The BCR is a membrane-bound form of the soluble immunoglobulin (antibody) that will be produced by that cell upon activation, as a consequence of the ability to bind a specific antigen. BCRs can bind to antigens in their native form, so that the B cell can detect unprocessed antigens. In contrast, the TCR is not secreted and stimulation via the TCR may lead to the activation of the T-cell response. Antigen recognition by T cells is also different from antigen recognition by B cells. T cells do not react to antigen in its unprocessed form, but rather recognize peptide fragments bound in the MHC of APCs (as described above; see also Fig. 21–1). These differences allow B and T cells to defend the host in 2 different ways: by directly recognizing the pathogen and by recognizing cells infected by the pathogen.
The TCR is composed of 2 protein chains joined together by disulfide bonds (Fig. 21–4A). This dimer can be composed of either α and β chains or γ and δ chains. The αβ TCR is present on the majority of mature T cells found in the blood and lymphoid organs while γδ TCR-bearing T cells are found primarily in skin and intestine. The TCR is expressed at the cell surface in a molecular complex that includes proteins that have intracellular signaling domains (immunoreceptor tyrosine activation motifs; ITAMs): CD3δε, CD3γε, and TCRζζ Once the TCR is bound by specific peptide/MHC complexes, the ITAMs in the CD3 and TCR molecules aggregate together and initiate an intracellular signaling cascade.
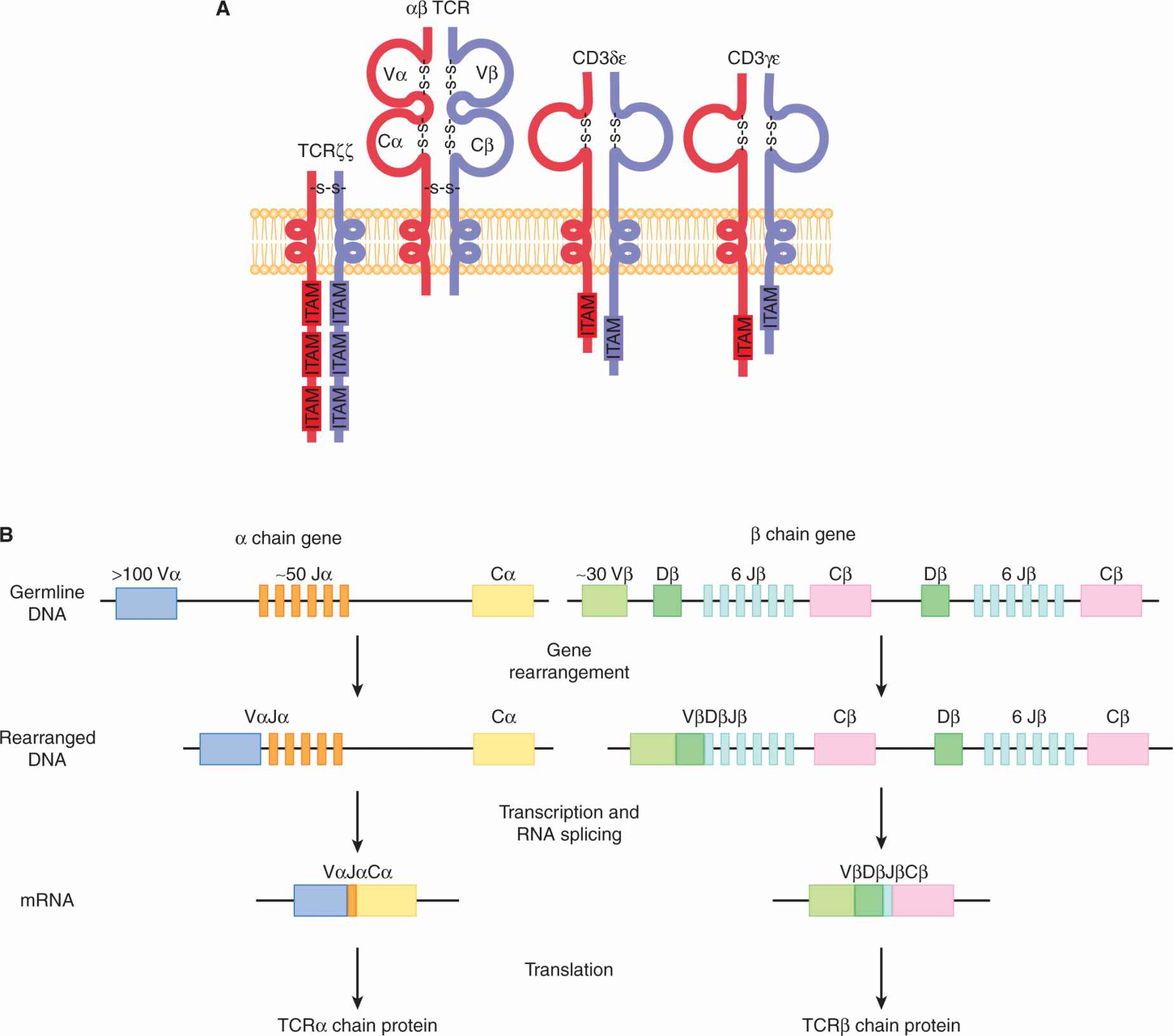
FIGURE 21–4 The TCR. A) The TCR is composed of 2 chains, α and β, each containing 2 immunoglobulin domains formed by an intrachain disulfide bond. The TCR is associated with the dimeric proteins CD3δε, CD3γε, and TCRζζ, and together with the αβ TCR, they comprise the TCR complex. ITAM, Immunoreceptor tyrosine-based activation motifs. B) The variable (V), diversity (D), joining (J), and constant (C) domains of the TCR are encoded by corresponding gene segments that undergo somatic rearrangement to generate the αβTCR heterodimer. In the mouse, the α chain consists of more than 100 V, approximately 50 J, and 1 C gene segments. The β chain consists of approximately 30 V regions and 2 clusters each of 1 D, 6 J, and 1 C gene segments. Following V(D)J gene rearrangement and transcription, the RNA is spliced to the C gene segment. The resulting messenger RNA (mRNA) encodes the TCR chains that dimerize to form the complete TCR protein.
The TCR chains are not encoded as single transcripts; rather, they are the result of genomic DNA rearrangements that bring together different gene segments and splice them into a combined exon (Davis, 1990; see Fig. 21–4B). This genomic splicing is dependent on an enzyme known as recombination activation gene recombinase (RAG recombinase) that is expressed exclusively in developing T and B cells (Krangel, 2009). The TCRα chain is comprised of 2 separate rearranged gene segments brought together during recombination. Variable (V) gene segments undergo rearrangement with joining (J) gene segments and are expressed together with constant (C) regions, forming the α chain of the TCR. The β chain has 3 rearranged gene segments: variable, diversity (D), and joining regions that undergo rearrangement and are expressed with the constant region of the β locus. The large number of possible combinations of VJ in the α chain and VDJ in the β chain results in enormous diversity among T-cell populations, especially as each mature T cell will rearrange its own TCR independently (Krangel, 2009; see Fig. 21–4B). In addition, a strategy called “N region diversity” occurs that generates additional nucleotides during the rearrangement process, which increases the repertoire of antigens that can be recognized by each TCR. The specificity of the TCR for a defined peptide bound in the groove of an MHC molecule is determined by regions of the TCR called complementary determining regions (CDRs). Interactions of the CDR with the peptide/MHC complex are a result of the combination of the V chains expressed, as well as the diversity generated by pairing of the VJ and VDJ gene segments.
21.3.2 T-cell Activation
The functional status of a mature T cell depends on whether it expresses a receptor that can bind to a peptide/MHC molecule expressed by a mature APC. T cells can exist in a naïve or resting state, or they can be effector T cells, which means that they have engaged a mature DC cell bearing their cognate antigen and have differentiated subsequently into functional T cells. Alternatively, they can be memory T cells, which are previously activated antigen-specific cells that persist after the pathogen has been eliminated. These 3 states (naïve, effector, and memory) impart quite different phenotypic and functional properties (Fig. 21–5). These stages of development have been studied most extensively in the CD8+ MHC class I-restricted lineage of T cells, and thus much of the following information will refer to CD8+ T cells; however, they also broadly apply to CD4+ T cells.
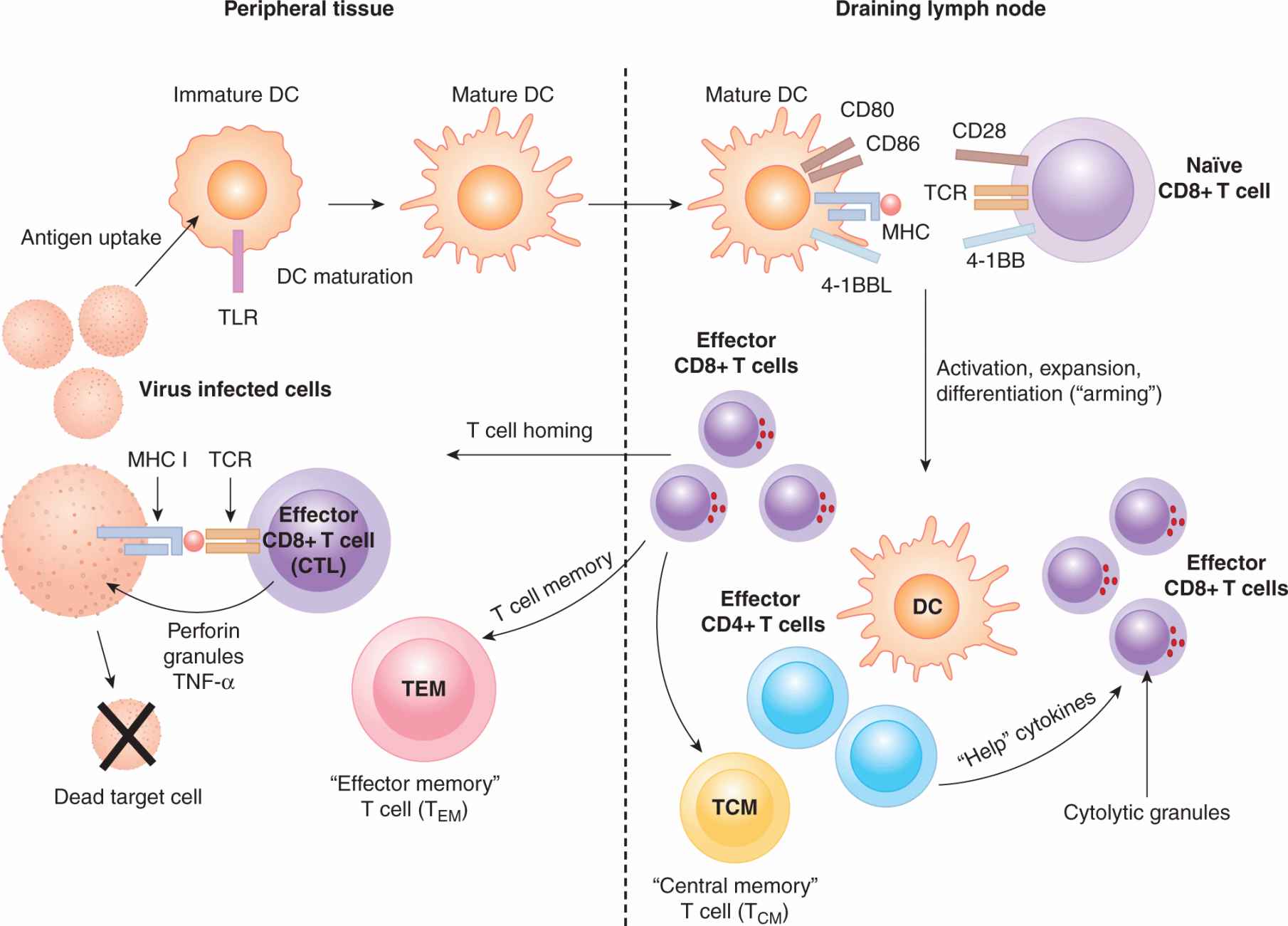
FIGURE 21–5 The stages of a cytotoxic T-cell response. CD8+ T-cell immune responses begin when a DC is activated by a pathogen or inflammatory signal in the periphery. Antigens are picked up by the DC and processed into peptides and placed onto MHC class I molecules via the cross-presentation pathway. Mature DCs migrate to T-cell areas in the lymph nodes, where they interact with naïve T cells. The T cell recognizes its cognate peptide bound to MHC class I on the DC, resulting in activation of the T cell. If the TCR signal is followed by costimulation by CD28 on the T cell interacting with CD80 and CD86 on the DC, the T cell will begin to proliferate, expanding the clone of the T cell that has specificity to the antigen. The CD8+ T cell will become armed by exposure to cytokines resulting in cytolytic granule formation. The T cell then exits the lymph node and traffics to sites of inflammation to engage and kill cells that present the cognate antigenic peptide on their MHC class I molecules.
Most T-cell responses can be subdivided into 3 phases: activation (also known as priming), expansion, and contraction (Fig. 21–6). A naïve T cell has a high threshold for activation, which means that when a mature T cell encounters its cognate antigen/MHC complex for the first time, it is slow to respond and requires several signals to initiate proliferation and acquisition of effector function. The DC is the main cell responsible for T-cell activation. During priming, a DC acquires antigen and matures at the site of infection and travels to the draining lymph node where it enters the T-cell area and interacts with the T cells resident there. An “integrin” receptor-ligand interaction occurs between leukocyte function-associated antigen (LFA)-1 and LFA-2 on the T cell and intercellular adhesion molecule (ICAM) expressed on the DC. These interactions initially slow migration of the DC past the naïve T cell and then help bring the 2 cell membranes into close proximity, allowing MHC/TCR interactions (Dustin and Cooper, 2000; Fig. 21–6). If a mature DC presents a peptide on its MHC molecules that is recognized by the specific TCR on the T cell, a signal will be sent into the T cell. This antigen-specific recognition, together with other receptor ligand interactions, promotes a functional T-cell response. One of the well-studied molecules that leads to optimal T-cell activation is CD28. The ligands of CD28 are CD80 (B7-1) and CD86 (B7-2), which are upregulated on the surface of mature DCs. This CD28 costimulatory signal results in the production of IL-2, a cytokine critical for T-cell proliferation and survival (Fig. 21–6). T-cell activation also results in the increased expression of several other molecules, including CD154, which is the ligand for the TNF family member CD40 that is present on the surface of the DC. The binding of CD40 on the DC results in the upregulation of costimulatory molecules, cytokines, and increased DC survival, allowing for prolonged antigen presentation (Quezada et al, 2004). The positive feedback loop between T cells and the DC results in expansion of antigen-specific T-cell clones. These expanded T cells are now functional effector T cells that change their chemokine and integrin expression patterns; they leave the lymph nodes and home to sites of inflammation.
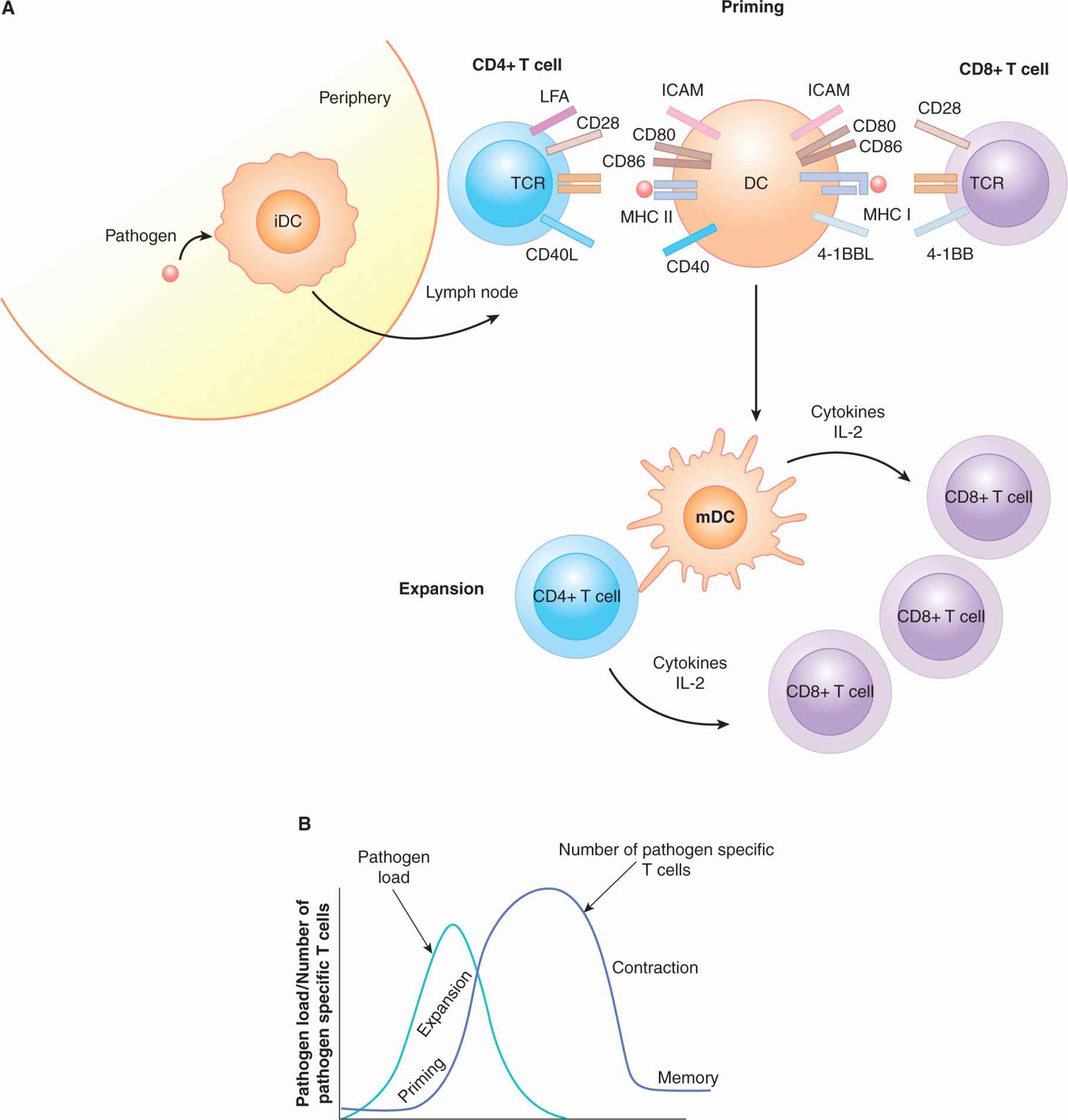
FIGURE 21–6 T-cell activation: priming, expansion, and contraction. A) After sensing tissue damage or infection in a peripheral tissue, the DC bearing antigen migrates to the T-cell area of a draining lymph node. DCs and T cells mingle in the lymph node, adhering to one another through interactions between LFA (lymphocyte function-associated antigen) on the T cell and ICAM (intercellular adhesion molecule) on the DC. A T cell with a TCR that is specific for the antigen obtained at the site of infection will interact with the DC and signal through the antigen-specific TCR. At the same time, CD28 on the T cell will bind to CD80 and CD86 on the DC. This signal is referred to as costimulation. CD4+ T cells will express CD40 ligand (CD154) that binds to CD40 on the DC, promoting the antigen-bearing DC’s survival and increasing cytokine production. Antigen-specific CD8+ and CD4+ T cells that receive these signals will then begin to divide. These expanded antigen-specific T cells then travel to the peripheral tissues to clear the infection. B) The T-cell immune response can be divided into several phases. Priming occurs when an antigen is first presented to naïve T cells and they are induced to expand for the first time. As the levels of antigen wane because of clearance by the immune system, contraction of the antigen-specific T-cell clones occurs. After contraction, a greater number of specific T cells remain than before the infection. These cells are referred to as memory T cells and have properties that allow them to react rapidly to reinfection with the same pathogen.
21.3.3 T-cell Memory
After the pathogen that initiated a T-cell response has been cleared, the effector T cells that responded to that pathogen disappear, leaving only a small pool of specialized T cells behind, known as memory T cells. These cells respond rapidly upon a re-encounter with the appropriate antigen, and thus offer protection from subsequent infections with the same pathogen. Memory T cells are CD8+ and can be subdivided into 2 categories: T effector memory (TEM) and T central memory (TCM) cells. These subsets can be defined by their localization, surface molecule expression, and function. The TCM cells are found in secondary lymphoid tissues. These cells express the chemokine receptor CCR7, express high levels of CD62L, and do not have preformed lytic granules. TCM cells most likely represent a pool of antigen-specific CD8+ T cells that are ready to expand again upon a second encounter with their cognate antigen. The TEM cells are found in nonlymphoid tissues: these cells have preformed cytolytic granules and are ready to act immediately at the sites of pathogen entry. TEM cells are identified by virtue of the profile CD8+, CD62 low, and CCR7–. The maintenance of memory CD8+ T cells is dependent in part on 2 cytokines, IL-7 and IL-15 (Kalia et al, 2006).
21.3.4 T-cell Subsets
CTLs are CD8+ T cells that possess the ability to kill target cells directly if the TCR is specific for peptides presented by MHC class I molecules expressed by the target cell. Differentiation of the CD8+ T cell from a resting naïve T cell into a mature CTL includes the formation of cytolytic granules (see Fig. 21–5). The cytolytic granules contain the molecules Perforin and proteases of the Granzyme family. After TCR engagement on the CTL, the cytolytic granules fuse quickly with the cell membrane, releasing Perforin and Granzymes. Perforin forms pores in the target cell membrane allowing the Granzyme proteases to enter. Granzymes initiate cleavage of proteins inside the target cell, and the result is apoptosis.
Th cells express the CD4 coreceptor and recognize peptides presented by MHC class II molecules. Th cells can be divided into several subgroups that are defined by their functional properties and/or their ability to produce a defined cytokine profile (Fig. 21–7). The first identified Th cell subsets were termed Th1 and Th2 and represented 2 discrete pathways of CD4+ mature T-cell differentiation. Th1 cells express the transcription factor T-BET (Szabo et al, 2000); they produce high levels of the cytokines IL-2, TNF-α, and IFN-γ, and induce IL-12 production by APCs. A Th1 cell augments primarily a CD8+ CTL-type response focused on elimination of intracellular pathogens, such as viruses and intracellular bacteria. Th2 cells mediate humoral (antibody) responses that are directed generally against extracellular pathogens and parasites. The Th2 T cell produces IL-4 among other cytokines: binding of IL-4 to its receptor on CD4+ T cells results in signal transducers and activators of transcription 6 (STAT6) activation and transcription of GATA3, which is the master transcription factor (see Chap. 8, Sec. 8.3.1) required for Th2 cell function (Zheng and Flavell, 1997).
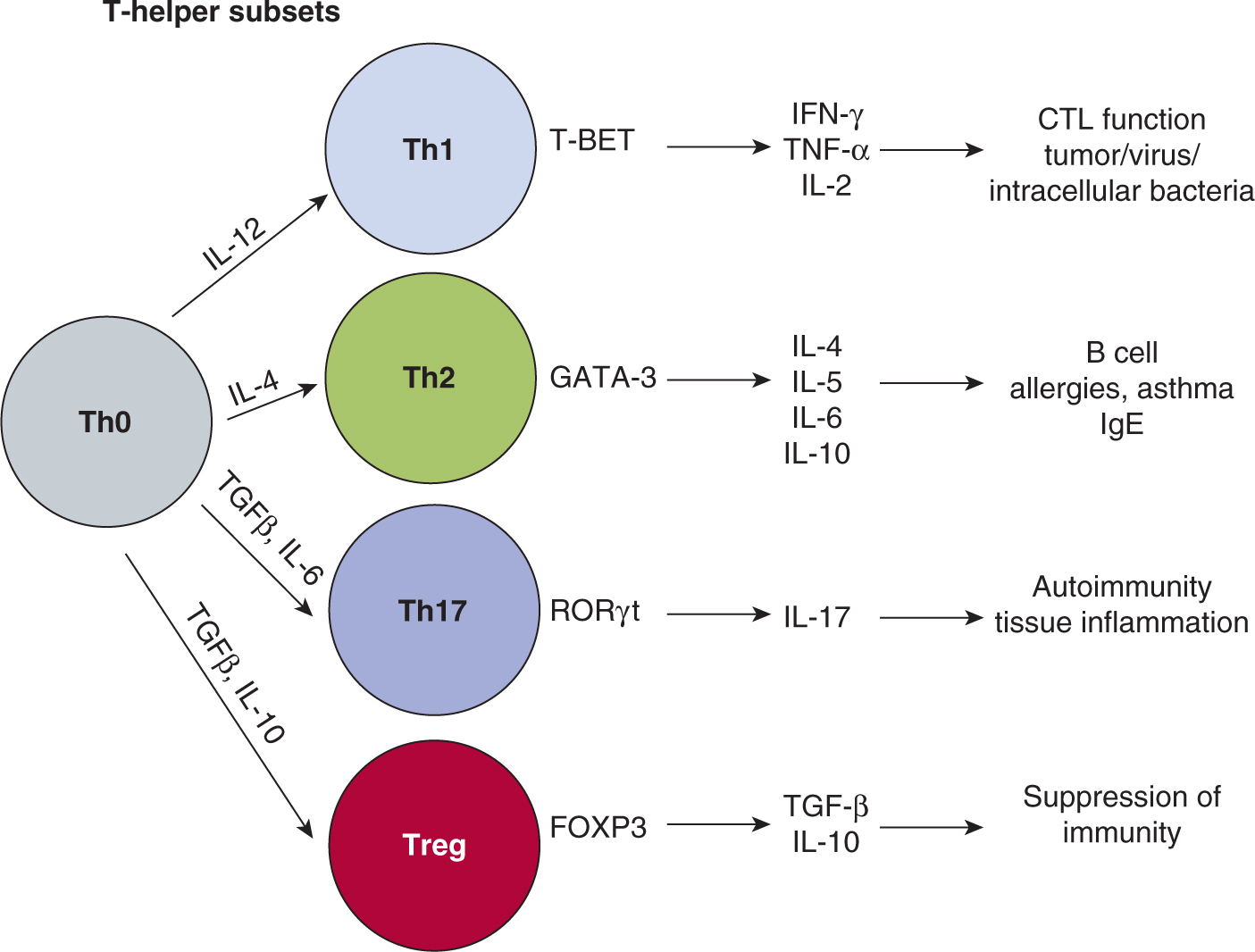
FIGURE 21–7 Th cell subsets. Th cells express the CD4 coreceptor and recognize peptides presented on MHC class II molecules of APCs. After activation through TCR engagement the T cell goes down 1 of 4 differentiation pathways, resulting in different effector functions. These pathways begin when a naïve CD4+ T cell (Th0) is activated in the presence of different cytokine and signaling conditions. The differentiation pathways have been identified to rely on transcriptional patterns enforced by specific transcription factors. After differentiation the subsets produce signature sets of cytokines that characterize their final effector function.
A newly identified Th subset is named Th17 for the ability to produce the cytokine IL-17, which is believed to act on local tissues to promote inflammation (McGeachy and Cua, 2008). These cells are required for many types of autoimmune diseases (Gutcher and Becher, 2007), and their possible role in antitumor responses is of great interest (Ji and Zhang, 2010).
Regulatory T cells (Tregs) represent another CD4+ T-cell population that has the ability to inhibit immune responses. This subset is key for preventing autoimmune diseases. In the mouse, they have been shown to express the markers CD25 and FoxP3, and their presence is dependent on the transcription factor FoxP3. There are 2 major sources of Tregs. One subset develops during thymic selection and is called natural T reg (nTreg). The second group of Tregs originates from mature peripheral CD4+ T cells that are converted into regulatory cells through signals in their environment; they are referred to as induced Tregs (iTregs) (Jonuleit and Schmitt, 2003; Josefowicz and Rudensky, 2009; Sakaguchi et al, 2008).
21.3.5 Suppression of T-cell Activation
Activation of T cells is required and desirable to resolve many infections, but uncontrolled T-cell activation may result in damage to the surrounding tissues. A balance between costimulatory activators and inhibitory molecules enables inhibition of a T-cell response when appropriate. Two of these inhibitory molecules are cytotoxic T-lymphocyte antigen (CTLA)-4 and programmed death (PD)-1. CTLA-4 is upregulated after TCR engagement on T cells, although it is constitutively found on Tregs. CTLA-4 competes with CD28 for binding with CD80 and CD86. Studies show that CTLA-4 has a higher affinity than CD28 for CD80 and CD86. Clustering of CD80/86 on the surface of APCs sends a signal to the APC to activate an enzyme called indoleamine 2,3-deoxygenase (IDO), which, in turn, metabolizes tryptophan and inhibits T-cell proliferation. Signaling through CTLA-4 into the T cell recruits the Src homology domain-containing phosphatase (SHP)-1 and interrupts the TCR signal. The critical role of CTLA-4 in inhibiting T-cell responses is evident in mice that have been genetically manipulated to lack the CTLA-4 protein (CTLA-4 “knockout” mice), as these mice develop severe autoimmunity (Fife and Bluestone, 2008).
PD-1, like CTLA-4, is expressed on activated CD4+ and CD8+ T cells, but is always present on Tregs. There are 2 known ligands for PD-1. PD-L1 (B7-H1) exhibits broad expression; it is found on many nonhematopoietic cells, as well as immune cells, including APCs. The expression of PD-L2 is restricted mainly to APCs. Expression of PD-L1 has been found on many tumors and correlates with poor prognosis. Signaling though PD-1 blocks the function and survival of effector T cells. Tregs have been shown to express both PD-1 and PD-L1 (Fife and Bluestone, 2008).
21.3.6 T-cell Tolerance
Because the rearrangements of the TCR are random, inevitably combinations will arise that are specific for self antigens. One of the main roles of the thymus is the production of functional, but not autoreactive T cells. This process can be broken down into 2 major events (Sebzda et al, 1999). First, a T cell must express a TCR that is able to interact with self MHC molecules. This process is known as positive selection and is important for selecting only those T cells that express receptors that recognize self MHC, as all foreign peptides are presented by self MHC. As a consequence of positive selection, each T cell in the repertoire is selected for recognition of the different MHC molecules in a given host; selection is based on weak interactions between the TCR and MHC presenting self peptide. Cells that essentially cannot bind to self MHC do not receive any signal through their TCR and undergo apoptosis (“no selection”). In contrast, T cells that have TCRs with high avidity to self peptides presented by the MHC are deleted; this process is referred to as “negative selection.” Alternatively, some high avidity, self-reactive thymocytes are rendered tolerant by a process called anergy, which results in T-cell inactivation. Finally, T cells with high avidity to self peptides may also differentiate into Tregs, which play a critical role in the maintenance of peripheral tolerance (Sakaguchi et al, 2003). A schematic of the model of thymocyte selection is shown in Figure 21–8.
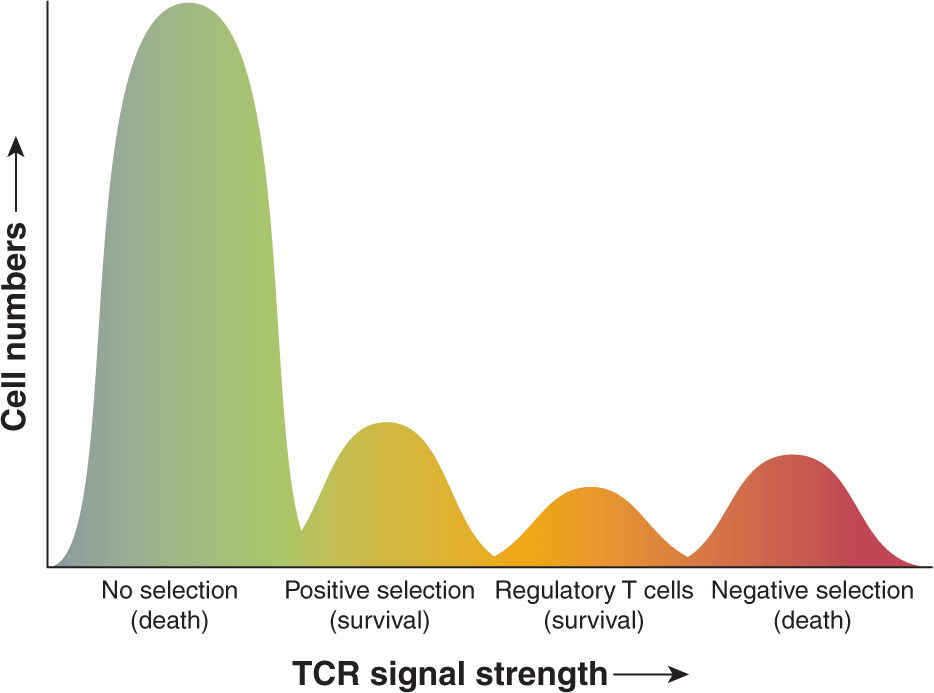
FIGURE 21–8 Thresholds of T-cell selection. T cells are selected during development in the thymus for their ability to bind peptides in the context of self-MHC molecules but at the same will not be activated by self-peptides to become effector cells. Random genomic re-arrangements of the TCR genes result in receptors with various affinities for self-peptides bound by MHC. T cells that essentially cannot bind to self-MHC do not receive any signal through their TCR and undergo apoptosis (“no selection”). Cells that bind self-MHC, but do not react strongly to self-peptides, finish development and enter the periphery as naïve T cells (“positive selection”). T cells with intermediate affinity to self-peptides will differentiate into regulatory T cells (Tregs). Finally, T cells that develop high-affinity TCRs, which would result in strong affinity to “self,” are deleted (“negative selection”).
Some autoreactive T cells escape negative selection in the thymus and enter the periphery. It is likely that this process occurs when some self antigens are not expressed in the thymus and therefore potentially self-reactive T cells cannot be rendered tolerant. The mechanisms involved in preventing these T cells with self-specificity from becoming activated either can be intrinsic to the T cell or a result of immune regulation by other cells.
Intrinsic mechanisms of peripheral tolerance include deletion and anergy, which are similar to the self-tolerance mechanisms that occur in the thymus. DCs have the ability to imprint the “fate” of deletion or anergy of mature self-reactive T cells. DCs can display self antigen that has been acquired from tissues and, if the DC is in a resting state, it renders T cells tolerant to these self antigens. However, as described in Section 21.2.2, DCs also have the ability to activate the immune system. The distinction that permits the induction of T-cell tolerance or T-cell immunity is the “functional status” of the DC: resting or steady state DCs present peptide/MHC complexes that can lead to induction of tolerance, whereas mature or activated DCs induce immunity (Steinman et al, 2003).
There are certain thresholds or concentrations of self antigen/MHC complexes that are required to induce tolerance (Ohashi, 1994). If tissue-specific antigens are not presented at an immunologically detectable level, T-cell ignorance is the result. In this situation, self-reactive T cells exist in the T-cell repertoire in a naïve or ignorant state. However, if these cells encounter antigen in an immune-stimulatory setting, these cells can be activated to destroy self-tissues. This scenario has been demonstrated experimentally in many models, including a mouse model of multiple sclerosis where myelin fragments are injected with a powerful immune stimulator (complete Freund adjuvant [CFA]). These myelin antigens are presented to naïve self-reactive T cells in the context of local inflammation induced by the CFA. The myelin-specific T cells then become activated and attack the myelin sheaths in the nervous system (Mendel et al, 1995).
Tregs are critical for the maintenance of peripheral immune tolerance in both mouse and man. Mice lacking Treg expression because of a mutation in the foxp3 gene (the transcription factor that is required for Tregs), succumb to severe multiorgan autoimmune disease. In humans, mutations impairing Treg development or function are correlated with the fatal autoimmune disease immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX; Wildin and Freitas, 2005). Current models propose that Tregs are antigen specific, but suppress in a nonspecific manner. This duality implies that to function properly they need to encounter self antigen and subsequently suppress the function of other T cells with various antigen specificities in their area. This phenomenon is known as “bystander suppression.” Tregs function in various ways, including cell contact- and soluble factor-mediated mechanisms. Activated Tregs possess the ability to kill directly both T cells and APCs in their local environment by producing Granzyme B that can enter surrounding cells and induce apoptosis (Gondek et al, 2005). This targeted elimination can reduce the quantity of activated T cells in an inflammatory site and terminate antigen presentation. Tregs have also been shown to inhibit killing by CTL by impairing granule exocytosis (Mempel et al, 2006). The high levels of CTLA-4 present on Tregs will bind to CD80 and CD86 on APCs resulting in increased intracellular levels of the tryptophan metabolizing enzyme IDO (Orabona et al, 2004). These high IDO levels then cause depletion of tryptophan from the local environment, thereby inhibiting T cell proliferation (Munn and Mellor, 2007). Tregs also express high levels of the IL-2 receptor CD25, and as a consequence, Tregs bind IL-2 more rapidly than other T cells in the local area. Thus, Tregs may function in part by limiting the amount of this T-cell growth factor available to effector cells nearby. Activated Tregs have also been shown to secrete the antiinflammatory cytokines IL-10 and transforming growth factor (TGF)-β (Sakaguchi et al, 2009); TGF-β also has antiproliferative functions and may contribute to limiting immune responses.
21.4 TUMOR IMMUNOLOGY
As described above, the immune system is equipped to recognize and eliminate foreign threats such as bacteria and viruses. However, studies demonstrate clearly that the immune system is also capable of recognizing tumors and mounting an immune response against our own “self” tissues. Many strategies that activate or enhance the immune response to tumors have been devised over the last few decades as therapies for cancer.
21.4.1 Tumor-Associated Antigens
One fundamental aspect that was critical to move the field of tumor immune therapy forward was the identification of tumor-associated antigens (TAAs). TAAs are intracellular or extracellular proteins that are expressed preferentially by tumor cells, but often can be expressed by normal cells. These proteins are processed and presented on MHC class I and II molecules in a similar manner to proteins of nontumor origin (eg, self proteins, or viral or bacterial proteins), as described above. Therefore, specific T cells can recognize TAAs through their TCRs. The identification of TAAs was important for tumor immunology and immunotherapy as it allowed for the detection and characterization of T cells that can recognize specific TAAs. It also allows for the design of immunotherapy that specifically activates TAA-specific T cells, for example by vaccination against specific TAAs.
21.4.1.1 Identification of Tumor-Associated Antigens The first TAA was identified by Boon and colleagues (van der Bruggen et al, 1991). Using a panel of CTL clones isolated from a patient with metastatic melanoma in combination with various sublines of melanoma tumor cells derived from the same patient, the authors identified a gene whose product was targeted by a particular CTL clone. This gene was not expressed in normal tissues. Furthermore, T cells from the original patient could recognize melanoma cell lines established from other human leukocyte antigen (HLA)-A–matched patients that also expressed the same TAA. Through this approach the MAGE, BAGE, and GAGE families of TAAs were identified.
Other approaches have successfully identified many TAAs. One method is referred to as SEREX (serological analysis of recombinant cDNA expression libraries; Sahin et al, 1995). Serum samples taken from cancer patients were postulated to contain antibodies that recognize TAAs. Expression libraries composed of complementary DNAs (cDNAs) isolated from autologous tumor cells were cloned into prokaryotic expression vectors. These libraries were used to bind antibodies and clones that were bound by antibodies were isolated and sequenced. The TAA recognized by the antibody was then identified based on the DNA sequence. Confirmation of the identified protein as a TAA was established in various ways, including evaluating protein expression in normal and malignant tissues. Important TAAs, such NY-ESO-1, were thus identified using the SEREX approach. Other methods to identify TAAs include the acid-elution of MHC-bound peptides from tumor cells and subsequent screening of peptide fractions for recognition by T cells from cancer patients, and, more recently, gene expression microarray analyses (see Chap. 2, Sec. 2.2.12) of tumor cells compared with normal cells. These approaches are depicted in Figure 21–9.
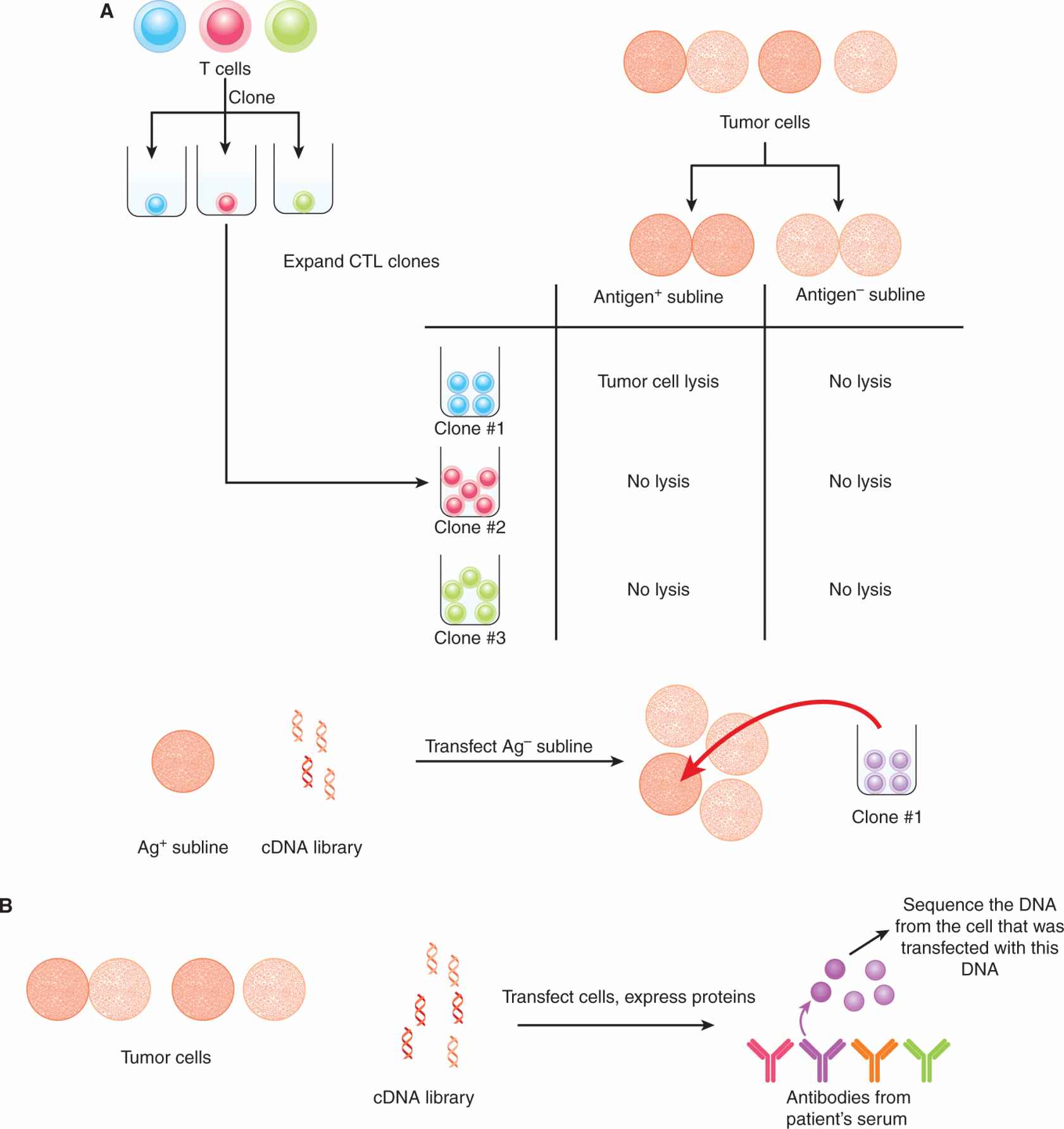
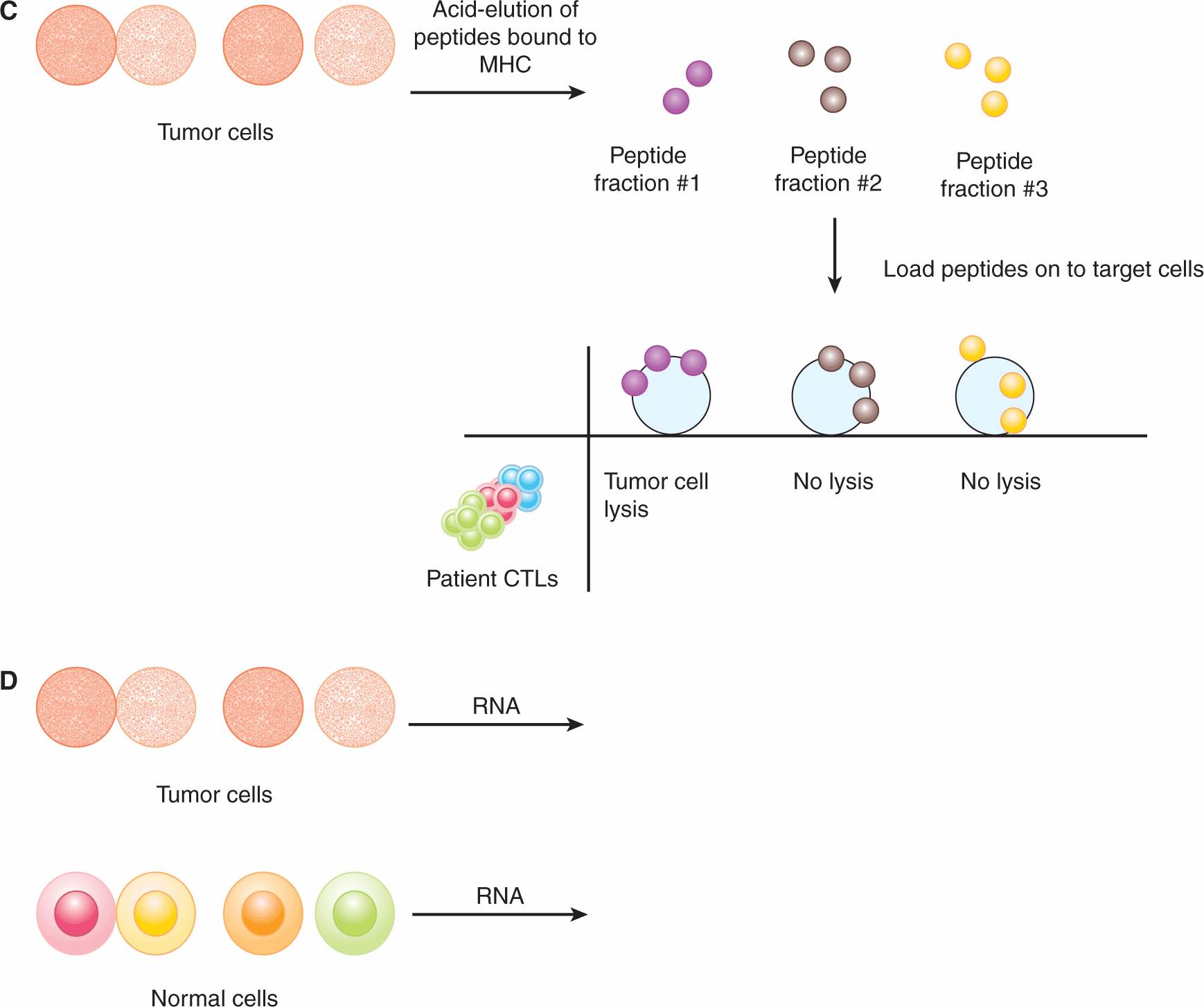
FIGURE 21–9 Approaches for identifying TAAs. A) CTLs from a cancer patient are cloned by plating them at 1 cell per well and expanding each clone. Autologous tumor cells are cultured in vitro and various sublines are obtained, some of which are “antigen-loss” variants. The CTL clones are tested for their ability to lyse the tumor cell sublines. If a clone is able to lyse one subline, but not another, the antigen expressed by the former is identified by transfecting antigen-negative target cells with cDNA libraries derived from the antigen-positive tumor cells. The DNA encoding the target antigen (the TAA) is isolated based on the ability of the CTL clone to lyse the target cell that was transfected with the TAA. B) In the SEREX approach, expression libraries composed of cDNAs isolated from autologous tumor cells are cloned into prokaryotic expression vectors. Serum samples from cancer patients (which should contain antibodies that recognize TAAs) are used to screen these libraries. Clones that are bound by antibodies are then isolated and sequenced. Other methods to identify TAAs include (C) the acid-elution of MHC-bound peptides from tumor cells and subsequent screening of peptide fractions for recognition by T cells from cancer patients, and (D) gene expression microarray analysis for upregulated RNA transcripts in tumor cells compared with normal cells.
Once a TAA has been identified, it is important to identify the peptide sequences in that protein that can be recognized by various TCRs. One common approach is to identify peptides that can be presented by HLA-A*0201 molecules and recognized by human T cells (Kawashima et al, 1998). HLA-A*0201 is prevalent in Caucasians and is often the focus of studies involving unique peptides that bind this HLA molecule. First, peptides that contained amino acid motifs that would be predicted to bind the HLA-A*0201 molecule were identified. The ability to bind HLA-A*0201 was verified using MHC-binding assays. These peptides were then tested for their ability to expand peptide-specific T cells from the peripheral blood T-cell population of HLA-A*0201-positive donors. These expansions were achieved by stimulating T cells with autologous DCs that had been exposed to the peptide of interest. After multiple rounds of peptide stimulation, the expanded T cells were tested for their ability to lyse peptide-coated target cells. Using this approach, multiple immunogenic peptides for the MAGE-A2, MAGE-A3, carcinoembryonic antigen (CEA), and human epidermal growth receptor (HER)-2/neu proteins were identified. A database of immunogenic TAA-derived peptides is available at www.cancerimmunity.org.
21.4.1.2 Types of Tumor-Associated Antigens TAAs can be classified into several broad categories: (a) mutation antigens, (b) cancer-testis antigens, (c) differentiation antigens, (d) overexpressed antigens, (e) viral antigens, and (f) antigens with unique posttranslational modifications (Fig. 21–10).
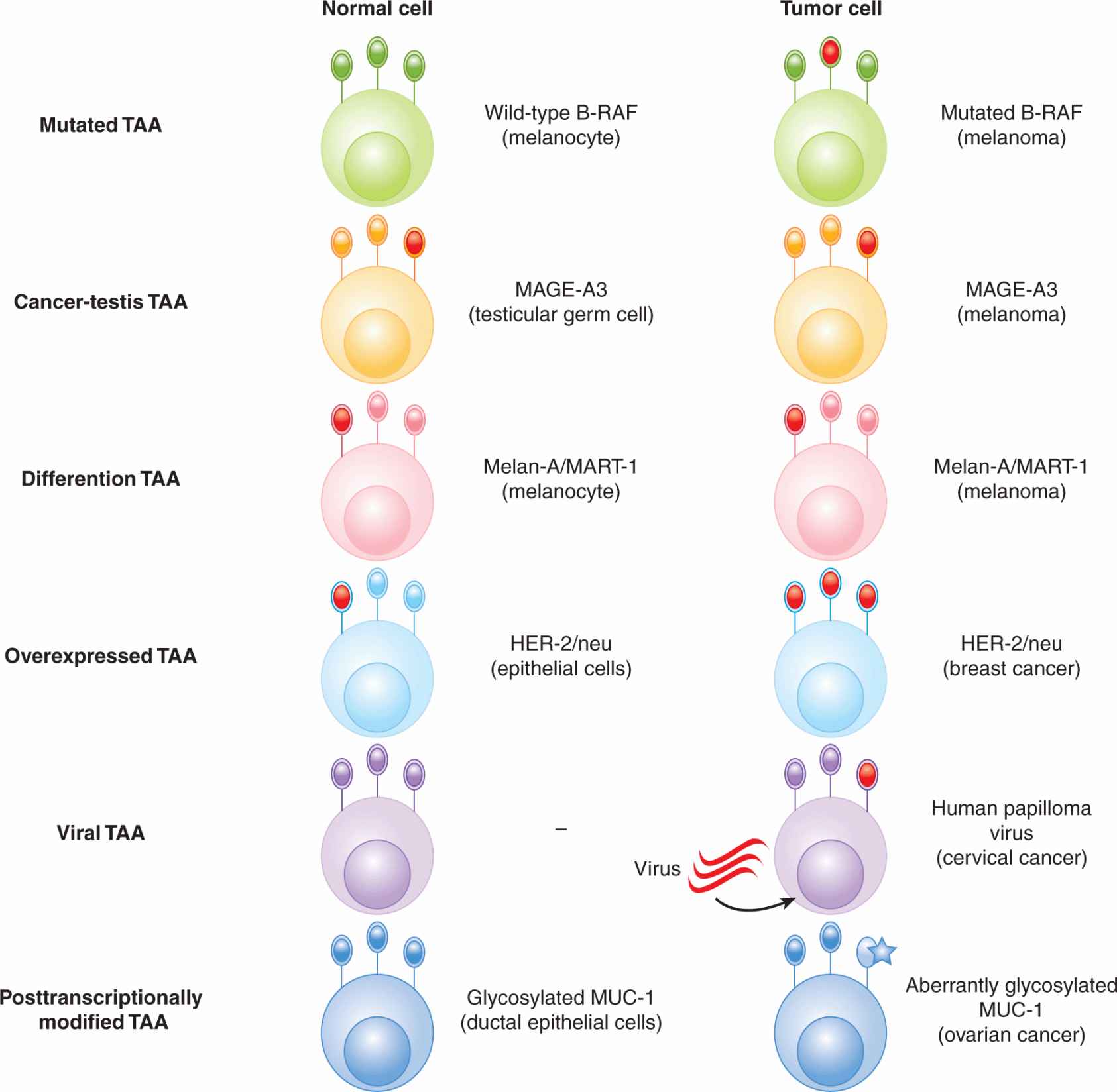
FIGURE 21–10 Types of tumor-associated antigens (TAAs). Various classes of TAAs are depicted and an example of each type of TAA is given. Expression on a normal cell and a tumor cell is shown. TAAs are depicted in red.
Mutation antigens are generally proteins that harbor mutations that are unique to an individual patient, although some mutations are common to a subset of patients. For example, approximately 2% of solid tumors and 40% to 60% of melanomas express a mutant form of the oncogenic protein B-RAF (see Chap. 7, Sec. 7.5.5). The overwhelming majority of patients have a valine → phenylalanine amino acid substitution at position 600. Mutated antigens are not necessarily recognized as unique by T cells, but they can serve as targets for other therapeutic approaches (eg, protein kinase inhibitors).
The expression of cancer-testis antigens is restricted to tumor cells and normal placental trophoblasts and testicular germ cells, and therefore represent appealing targets for immunotherapy. Cancer-testis antigens include the MAGE, BAGE, and GAGE proteins, as well as the commonly expressed NY-ESO-1.
Differentiation antigens are proteins that are expressed on both normal tissues and tumors derived from a particular tissue. For example, Melan-A/MART-1 is expressed on normal melanocytes, as well as malignant melanoma. CEA is expressed on normal colonic epithelium, as well as many carcinomas of the gut. Other differentiation antigens include gp100 (melanoma), tyrosinase (melanoma), and prostate-specific antigen (PSA-prostate cancer). Differentiation antigens also include oncofetal antigens, such as α-fetoprotein, which are expressed in various tissues during fetal development and are reexpressed on tumors.
Overexpressed TAAs are expressed on various normal tissues, as well as tumor cells but with higher expression on tumor cells. For example, Wilms tumor-1 (WT-1) is expressed in a proportion of breast cancers, lung cancers, and other tumor cells.
Viral TAAs are expressed by tumors that are induced by viral transformation. This includes Epstein-Barr virus (EBV)-induced tumors such as some lymphomas and nasopharyngeal carcinoma, and human papilloma virus (HPV)-induced cervical neoplasias (see Chap. 6).
Another class of TAAs consists of proteins where posttranslational modification occurs differently in normal cells compared with tumor cells. T cells are able to distinguish between the same peptide with different posttranslational modifications, such as differential glycosylation levels. For example, mucin-1 (MUC-1) proteins in tumor cells are generally hypoglycosylated in tumor cells compared with normal cells. Furthermore, the sugar moieties on tumor-associated MUC-1 tend to be shorter.
21.4.2 Tumor-Associated Antigen-Specific T cells in Humans
The development of MHC/peptide multimer reagents has enabled the direct detection of T cells specific for various TAAs. This reagent generally consists of soluble MHC class I molecules (eg, HLA-A*0201) folded together with a peptide of interest (eg, a peptide derived from a tumor-associated protein that is known to bind that particular HLA allele) (Fig. 21–11). Each MHC molecule is tagged covalently with a molecule of biotin. Multiple MHC/peptide complexes are then aggregated together using streptavidin, which possesses 4 biotin-binding sites. The multimeric nature of the reagent allows for higher avidity binding of specific TCRs, and any bound cells can be detected by flow cytometric analysis of the fluorochrome-conjugated streptavidin moiety. The first reported multimers were synthesized using HIV and influenza A-derived peptides (Altman et al, 1996). Although MHC/peptide multimers have also been constructed using MHC class II molecules and class II-binding peptides, they are generally not as robust as MHC class I-derived multimers (Vollers and Stern, 2008).
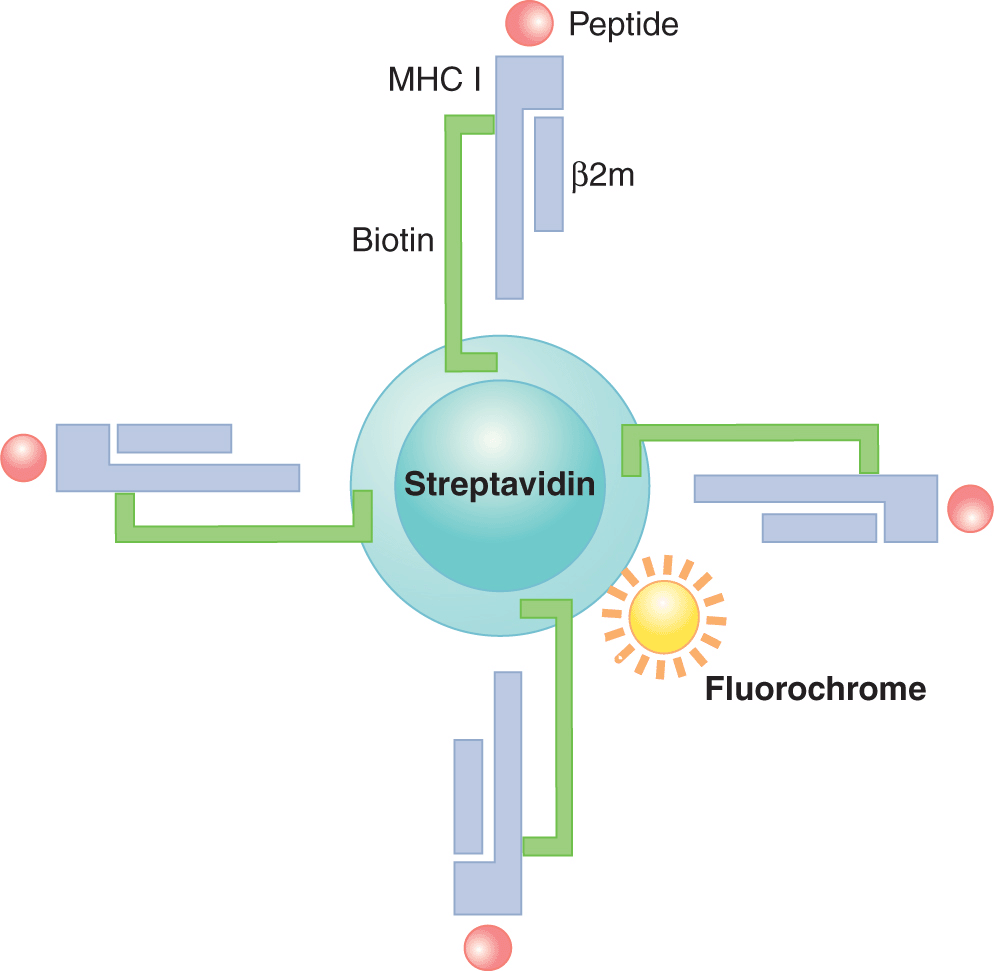
FIGURE 21–11 Structure of an MHC/peptide multimer. An MHC/peptide tetramer is depicted, where 4 MHC class I/peptide complexes are complexed together. Each MHC class I molecule is biotinylated; each biotinylated MHC class I/peptide complex is bound via the biotin molecule to a streptavidin molecule, and the streptavidin molecule is conjugated to a fluorochrome. These reagents are helpful for monitoring tumor specific T-cell responses.
Early studies using HLA-A*0201 MHC/peptide multimers were able to identify the presence of peripheral T cells that could recognize melanoma-associated antigens. These tumor-specific T cells could be detected directly ex vivo, without the need for in vitro stimulation or expansion using specific peptides. T cells specific for the melanoma-associated antigens Melan-A/MART-1 and tyrosinase were detected by ex vivo staining of tumor-infiltrated lymph nodes (Romero et al, 1998). MHC/peptide multimers were also used to isolate specific T cells by fluorescence-activated cell sorting (FACS), and these cells were found to be functional, as they could lyse autologous melanoma tumor cells in vitro (Romero et al, 1998).
The presence and function of tumor-specific T cells have also been demonstrated for other cancers. Numerous studies have demonstrated that some women with breast cancer have preexisting immunity against breast TAAs. In one study, 7 of 13 breast cancer patients had detectable tumor-specific T-cell responses at the time of diagnosis (Rentzsch et al, 2003). This response was demonstrated by evaluating the induction of IFN-γ RNA transcripts in response to stimulation with peptides derived from several common antigens expressed by breast cancers: MUC-1, HER-2/neu, CEA, NY-ESO-1, and SSX-2. HER-2/neu is expressed by a variety of cancers and preexisting immunity to HER-2/neu has been demonstrated in patients whose cancers expressed HER-2/neu (breast, ovarian, lung, colorectal, prostate) (Sotiropoulou et al, 2003). After a brief restimulation of peripheral blood T cells ex vivo with HER-2/neu peptide-exposed DCs, effector function of the restimulated T cells was evaluated by enzyme-linked immunospot (ELISPOT) assay (production of IFN-γ on a per-cell basis) and CTL assay (ability to kill target cells).
Clearly, immune tolerance against tumors is incomplete, as TAA-specific T cells can be detected in cancer patients. Whether these tumor-specific T cells can mediate antitumor activity, and how they might be induced to do so by therapeutic intervention, is a major challenge for successful development of immunotherapy.
21.4.3 Immune Surveillance
Historically, the concept of “immune surveillance,” where the immune system surveys the body and targets tumor cells even in the absence of therapeutic intervention, has not always been accepted. Early studies that argued against immune surveillance examined tumor development in mice that carried the “nude” mutation and lacked a thymus and were considered immunodeficient (Stutman, 1974, 1979). It was found that nude mice developed both carcinogen (methylcholanthrene [MCA])-induced and spontaneous tumors at similar frequencies to wild-type mice, and it was concluded that the immune system does not play a role in inhibiting tumor progression. Several factors that could influence the interpretation of this model have been identified. Firstly, it is now known that nude mice still have some intact immune cells. Secondly, the importance of the innate immune system (which is intact in nude mice) in antitumor immunity has been established. Thirdly, the genetic background of the nude mice used in the above studies (CBA/H background) express a particular isoform of the enzyme that metabolizes MCA. Therefore, nude mice could be more resistant to MCA-induced tumor formation, which could mask any consequences related to an impaired immune response.
Numerous studies provide strong evidence in support of cancer immunosurveillance (Dunn et al, 2004). That the immune system eliminates tumor cells was shown using various immunodeficient mice: (a) mice deficient in the Rag protein and therefore lacking all T and B cells, (b) mice deficient in the receptor for the immunostimulatory cytokine interferon-γ (IFN-γ receptor 1 knockout mice), and (c) mice deficient in the key intracellular signaling molecule downstream of IFN-γ receptor signaling (Statl knockout mice). These immunodeficient mice developed MCA-induced and spontaneous tumors at a higher frequency and with faster kinetics compared with immunocompetent mice (Kaplan et al, 1998; Shankaran et al, 2001). A model has been proposed to describe the role of the immune response during various stages of tumor progression, which includes three phases: elimination, equilibrium, and escape (Dunn et al, 2004) (Fig. 21–12).
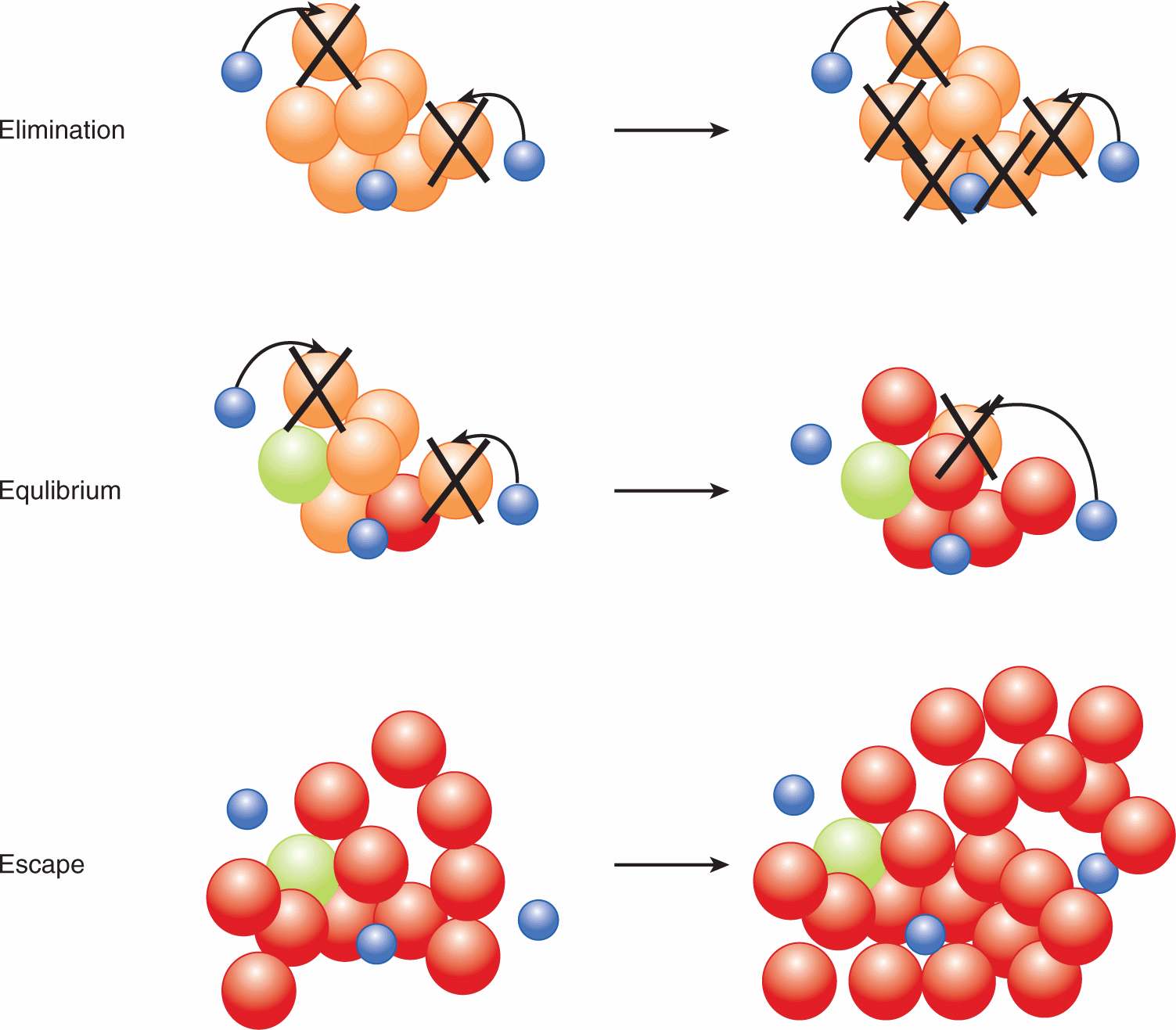
FIGURE 21–12 The three Es of cancer immunoediting. In the model proposed by Dunn et al (2004), immunoediting is comprised of 3 main phases. During the elimination phase, the immune system recognizes tumor cells and mediates their elimination. During the equilibrium phase, the immune system and the tumor are in dynamic equilibrium, where the combination of genomic instability of tumor cells and selective pressure exerted by the antitumor immune response results in editing, or “sculpting” of the tumor cell population. The elimination of certain tumor cells by the immune system results in the selection of less immunogenic tumors. During the escape phase, enough tumor cell variants arise that the tumor can escape elimination by the immune system. Blue, immune cell; orange/green/red, tumor cells.
During the elimination phase of immunosurveillance, the immune system recognizes tumor cells and mounts a response against the tumor. During the equilibrium phase, the immune system and the tumor are in dynamic equilibrium, where the combination of the genomic instability of tumor cells and selective pressure exerted by the antitumor immune response results in editing, or “sculpting” of the tumor cell population. The elimination of certain tumor cells by the immune system results in the selection of less immunogenic tumors. During the escape phase, enough tumor cell variants arise that the tumor can escape elimination by the immune system. Escape may be partly a result of reduced immunogenicity of tumor cells, such as reduced expression of TAAs, or downregulation of MHC class I, but there are many mechanisms whereby the tumor can evade the immune system. These mechanisms include negative regulatory immune cell types, immunosuppressive factors produced by tumor cells, and surface receptors that dampen the T-cell response.
21.4.4 T-cell Infiltration and Disease Prognosis
The importance of T-cell immunity in reducing clinical tumor burden in humans is highlighted in part by studies evaluating tumor tissues for infiltration by immune cells. In many studies where immunohistochemical or gene expression analyses of tumor tissue are performed, the data show an association between immune cell (especially T-cell) infiltration and better prognosis (eg, Galon et al, 2006; Tuthill et al, 2002; Zhang et al, 2003).
In an example of this type of study, genomic analysis for RNA expression was performed for 75 colorectal tumors (Galon et al, 2006). Expression levels of a cluster of Th1-related genes showed a statistically significant inverse correlation with tumor recurrence, thus implicating the Th1 response in dampening progression of human tumors. In the same study, immunohistochemistry for immune cell infiltration in paraffin-embedded tissue microarrays constructed from 415 colorectal tumors was also performed. High densities of CD3+ T cells, CD8+ T cells, Granzyme B (a cytotoxic granule produced by lytic T cells), and CD45RO (a T-cell activation marker) were found in patients without recurrence compared to patients with recurrence. In a multivariate analysis, the density of CD3+ cells in tumors was shown to be an independent prognostic factor, and these T-cell markers proved to be a better predictor of patient survival than standard histopathologically based staging methods.
21.4.5 Barriers That Inhibit Antitumor Immunity
Despite the evidence that the immune system can respond to tumor cells, it is difficult to estimate the relative importance of antitumor immunity in tumor progression. However, it is clear that when tumors do develop, the immune response is unable to control tumor growth. Various mechanisms can lead to failure of immune-mediated tumor control; many of them are the same mechanisms that prevent autoimmunity and thereby suppress immune responses against self-tissues. Various mechanisms of peripheral T-cell tolerance are discussed above (see Sec. 21.3.7): T-cell deletion, T-cell anergy, and the function of negative regulatory cells and molecules have all been demonstrated to possess a negative impact on the immune response to tumors in various models. In addition, factors produced by tumor cells, or by tumor-infiltrating immune cells are known to suppress antitumor T-cell responses. Two examples of tumor-related immunosuppressive factors are TGF-β and IDO.
TGF-β is produced by tumor-associated macrophages, tumor cells, and Tregs. It exerts immunosuppressive effects on multiple cell types, including DCs and effector T cells. TGF-β signaling in DCs results in the downregulation of MHC class II, CD40, CD80, and CD86, as well as the inhibition of the proinflammatory cytokines IL-12, interferon (IFN)-α and tumor necrosis factor (TNF)-α. Thus, TGF-β induces a tolerogenic phenotype in DCs. These tolerogenic DCs themselves have been shown to secrete TGF-β leading to the induction of Tregs in mouse and human models. The immune suppressive effect of TGF-β on T cells is also well-established. TGF-β signaling results in suppression of Granzyme A, Granzyme B, and FAS ligand in CTLs. In addition, iTregs can be generated by TGF-β.
Clinical trials for various agents aimed at blocking the effect of TGF-β have been conducted (Flavell et al, 2010). Blocking strategies include small molecule inhibitors, delivery of anti-sense molecules, and the use of blocking antibodies. Clinical responses have been limited, but further investigation into the immunological effect of these strategies and combination of TGF-β blockade with other immunotherapeutic strategies might lead to improved responses.
IDO is an important immunosuppressive mediator (Munn and Mellor, 2007). IDO is an enzyme involved in oxidative catabolism of tryptophan, and since tryptophan is important for T-cell proliferation and activation, depletion of tryptophan by IDO inhibits T-cell responses. IDO-secreting DCs have been found in patients with various cancers, including melanoma, breast cancer, and colon cancer (Munn et al, 2002). IDO-secreting DCs have also been found in tumor-draining lymph nodes and, furthermore, these IDO-secreting DCs can induce anergy of tumor-specific T cells in vivo. Immunohistological detection of IDO in tumor-draining lymph nodes of melanoma patients revealed that IDO expression correlated with poor prognosis (Lee et al, 2005; Munn et al, 2004). Tumor cells can also secrete IDO.
Understanding the mechanisms by which tumor-specific T cells fail to control tumor growth may form the basis for designing therapeutic strategies. For example, unresponsive tumor-specific T cells may need to be rescued ex vivo and then reinfused; negative regulatory molecules may need to be blocked; or deleted tumor-specific T cells may need to be replenished. Immunosuppressive molecules present in the tumor microenvironment may also need to be inhibited. It is likely that a combination of these approaches will lead to the most effective approaches to immunotherapy.
21.5 IMMUNOTHERAPY
21.5.1 Introduction to Immunotherapeutic Approaches
Our improved understanding of tumor immunology has led to a growing list of immunotherapeutic agents being approved for use against cancer. One broad category of agents consists of monoclonal antibodies (mAbs). Although the mechanisms of action of various mAbs are not always related to the antitumor T-cell response that is discussed above—many function in a manner completely independent of T cells—they are, nevertheless, important players in cancer therapy.
Immunotherapy that is aimed at augmenting antitumor T-cell immunity can be broadly categorized into (a) nonspecific immunotherapy, (b) specific immunotherapy, and (c) adoptive cell therapy. Each category encompasses many different strategies—some investigational and some approved for therapy. The small number of these strategies selected for discussion in this chapter are listed in Table 21–1.
TABLE 21–1 Selected approaches to cancer immunotherapy.
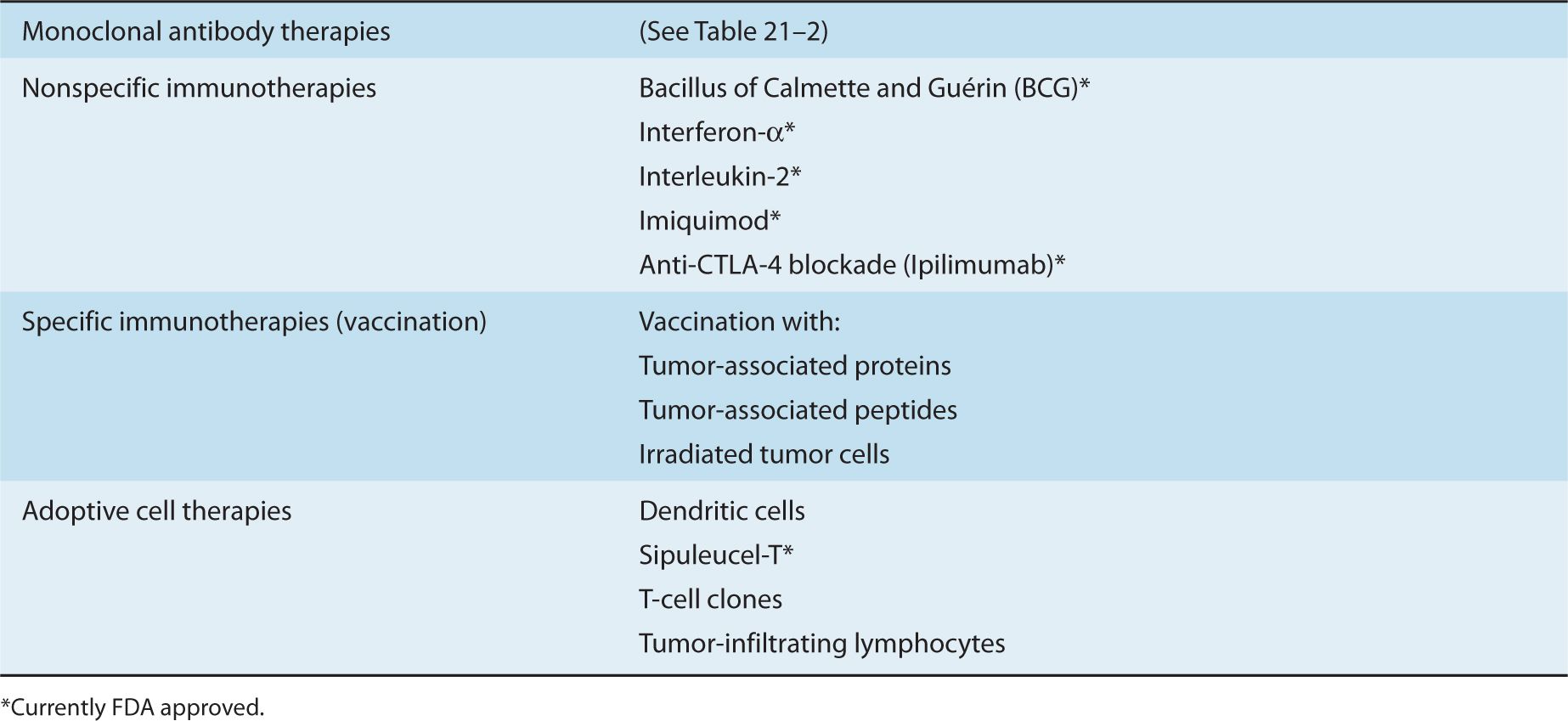
Nonspecific immunotherapy refers to strategies that augment general T-cell responses, in a “nonspecific” or “polyclonal” manner. Nonspecific immunotherapy includes the use of cytokines (eg, INFα, IL-2), immunological adjuvants (Imiquimod), and agents that target immunomodulatory molecules (anti–CTLA-4 antibody). Specific immuno-therapy focuses on the activation and enhancement of the number of T cells that can recognize TAAs by using vaccine strategies. Although the induction of an immune response by vaccination often refers to a prophylactic strategy, such as that used to limit viral infection, vaccination may also refer to therapy aimed at eliciting antitumor immune responses to eliminate established cancers. The field of prophylactic cancer vaccines will not be addressed in this chapter but is reviewed in Lollini et al (2006). Common vaccine approaches include the use of TAAs or peptides derived from TAAs. Other approaches are based on vaccination using whole tumor cells (autologous or allogeneic) that have been irradiated prior to infusion to prevent their proliferation following infusion. In adoptive cell therapy, autologous immune cells such as DCs and T cells are manipulated ex vivo and then reinfused.
21.5.2 Monoclonal Antibodies for Cancer Therapy
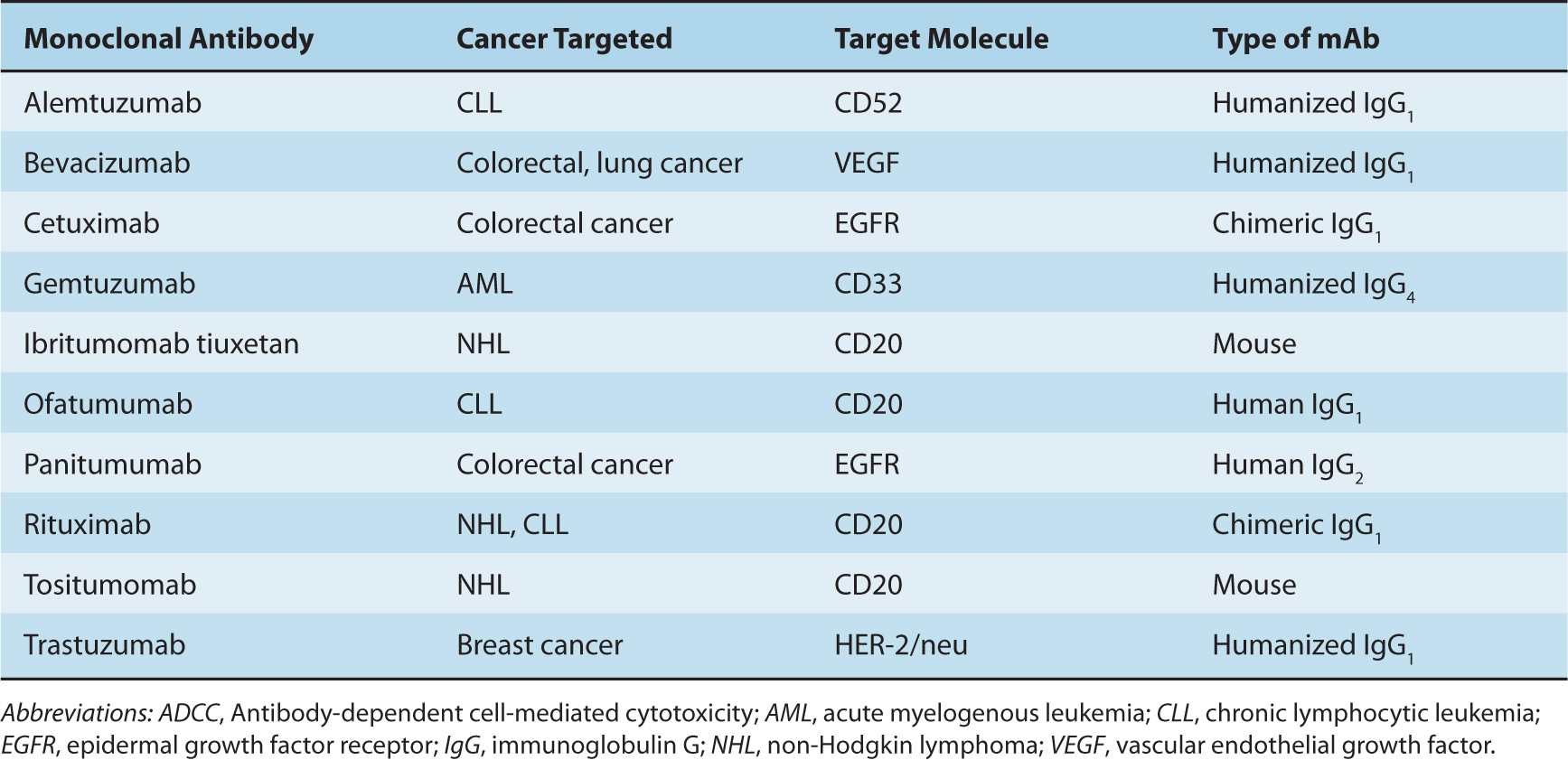
Monoclonal antibodies (mAbs) bind specifically to their target protein and can block the target protein’s function, trigger signaling downstream of the target protein, or deliver conjugated toxins to cells expressing the target protein. Current Food and Drug Administration (FDA)-approved mAbs include those that target antigens expressed by tumor cells (eg, CD20 on non-Hodgkin lymphoma and chronic lymphocytic leukemia), molecules that promote tumor growth (eg, epidermal growth factor receptor), or angiogenic molecules (eg, vascular endothelial growth factor). Table 21–2 lists examples of antibodies approved for cancer therapy.
TABLE 21–2 Table of monoclonal antibodies approved for cancer therapy.
21.5.2.1 Production of Monoclonal Antibodies A mAb that recognizes a given protein target is derived from an antibody molecule that was produced by a single B cell. Originally, mAbs were produced by fusing B cells from the spleen of an animal that had been immunized with the target protein, with a myeloma cell line that was selected for the inability to produce immunoglobulin. Primary spleen cells cannot survive ex vivo in unsupplemented culture medium. The myeloma cell line was also selected for a deficiency in an enzyme (hypoxanthine-guanine phosphoribosyltransferase [HGPRT]) that renders the cells unable to grow in culture medium containing hypoxanthine, aminopterin, and thymidine (HAT). Successful cell fusions between spleen cells (expressing HGPRT) and myeloma cells could therefore be selected by culturing in HAT-containing medium. Fused cells were then cloned: Cells were plated at limiting numbers and individual cells were expanded separately. These fused cells (called hybridomas) were then screened for secretion of antibodies that could bind the target protein of interest. Once a hybridoma with the desired antibody specificity is identified, large batches of antibodies can be produced by expanding the hybridoma in culture, or expanding the hybridoma in the peritoneal cavity of animals (mice, rabbits) and harvesting the ascites. A schematic of this classical method of antibody production, and an example of a current method based on a genetic approach, described below, are depicted in Figure 21–13.
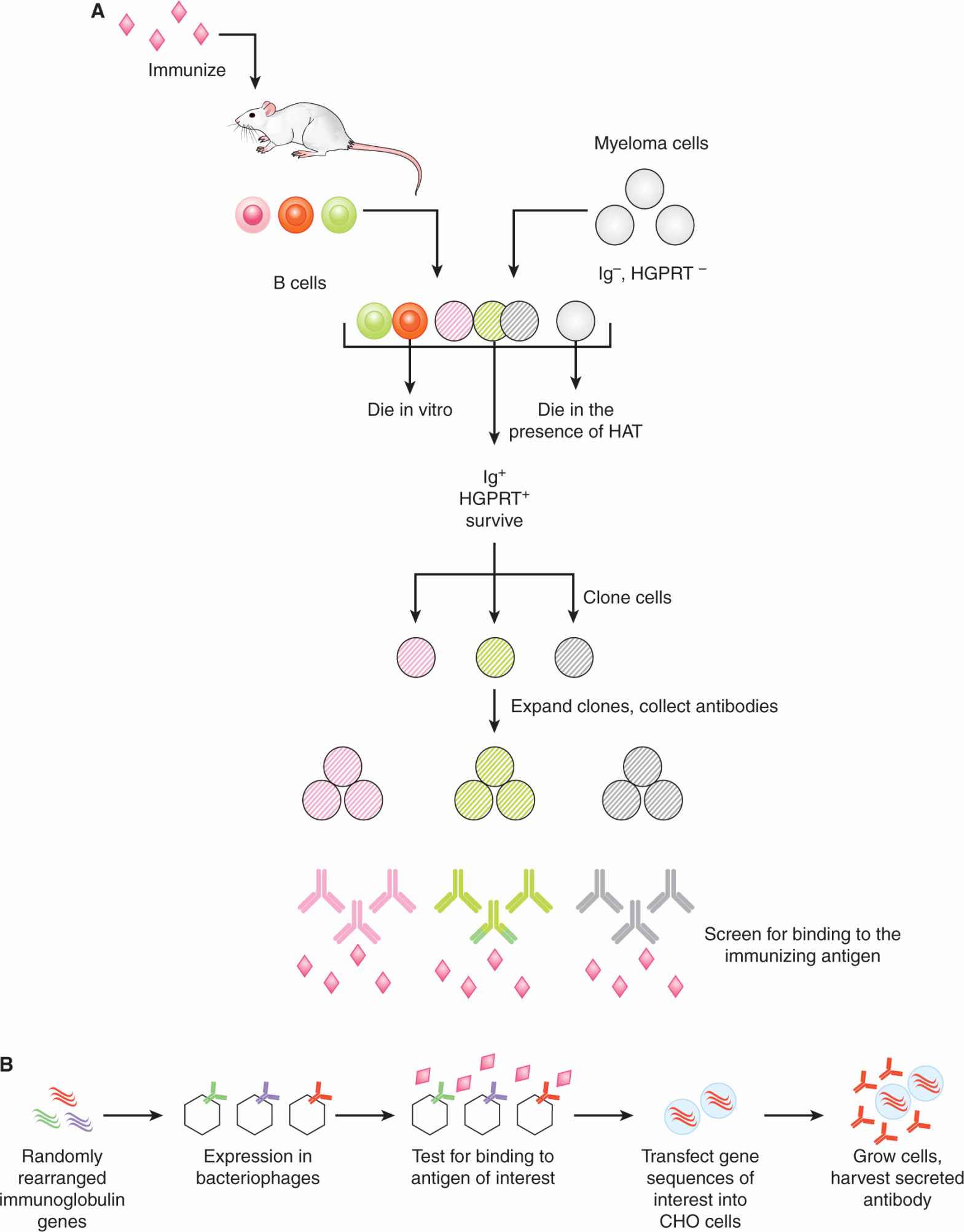
FIGURE 21–13 Methods of monoclonal antibody production. A) mAbs were initially produced by the fusion of B cells with a specialized myeloma cell line. Fused cells were cloned and then the clone that produced the antibody of interest was selected for antibody production. Further details are provided in the text. B) Other methods are based on recombinant technologies, where the desired antibody genes are transfected into cells such as Chinese hamster ovary (CHO) cells and antibodies are harvested from culture supernatants. In this example, the antibody genes of interest are identified by screening bacteriophages for binding to the antigen of interest. The bacteriophages have been engineered to express antibodies that are the product of random rearrangement of immunoglobulin genes.
21.5.2.2 Types of Therapeutic Antibodies Antibodies that recognize human proteins were generated initially by immunization of animals (eg, mice) with human proteins and isolation of specific antibodies, as described in the hybridoma approach above. However, mouse antibodies have limited therapeutic value for several reasons. First, mouse Fc domains (which is the “constant” domain of an antibody that is conserved amongst all antibodies of a particular class) are not recognized as efficiently as human Fc domains by human immune cells, and such recognition is required for some of the mechanisms of action of antibody therapy, described below. Also, mouse antibodies are recognized as foreign proteins by the human immune system and thus repetitive antibody administration would result in an immune response against the therapeutic antibody. One solution has been the generation of chimeric antibodies, which are engineered to contain the variable (Fv) region from a mouse antibody and the human constant (Fc) regions. An alternate approach is to generate humanized antibodies where the hypervariable regions of the variable antigen-binding domain (Fv) is derived from a mouse antibody and the rest of the Fv and the entire Fc region is derived from human sequences. To completely circumvent neutralizing antibodies with xenogeneic sequences, many current therapeutic antibodies are fully human. These antibodies can be isolated from mice that have been engineered to express only human antibodies, or they can be isolated by a phage display approach, where libraries of random recombinations of human antibody genes are expressed in bacteriophages. The antibody-expressing phages are then screened for the ability to bind the target protein. Recombinant antibodies are often produced by transfecting Chinese hamster ovary (CHO) cells with the desired antibody genes, as CHO cells grow well in suspension culture. Figure 21–14 illustrates these types of antibodies.
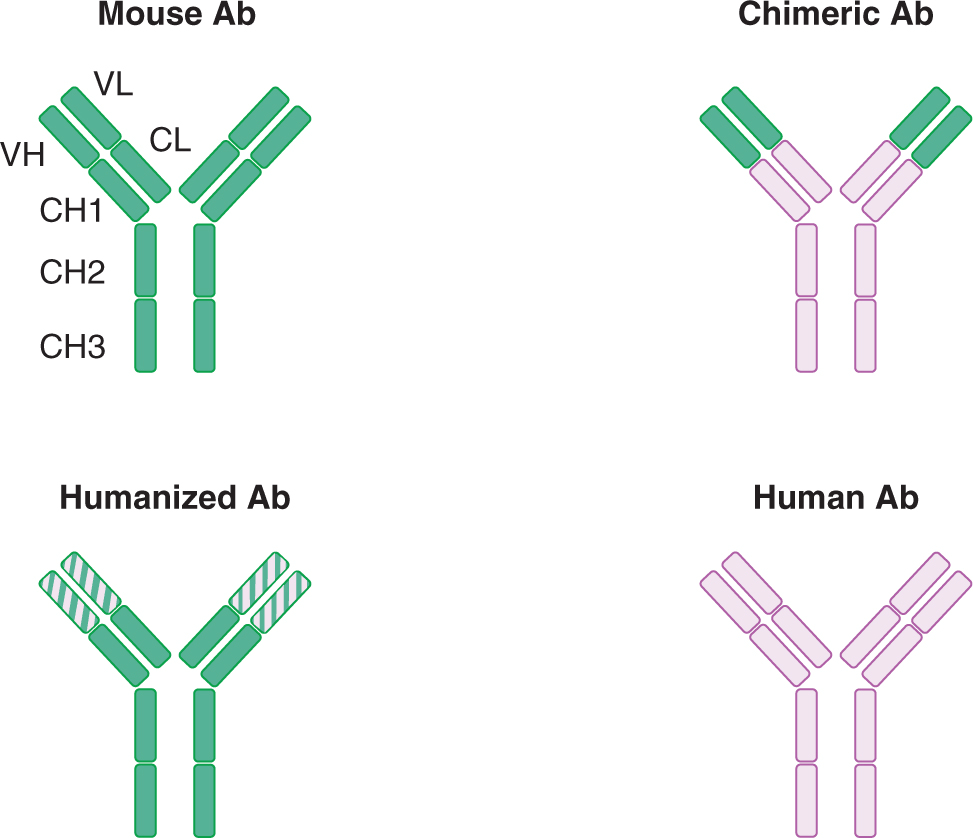
FIGURE 21–14 Chimeric and humanized antibodies. In chimeric antibodies, the variable fragment (Fv) of the antibody molecule is of mouse origin and the constant regions (Fc) is of human origin. In humanized antibodies, only the hypervariable region of the Fv is of mouse origin; the rest of the antibody molecule is of human origin.
21.5.2.3 Mechanisms of Action of Antibodies Antibodies that bind to receptors on the cell surface can block receptor signaling (by steric blockade of ligand binding) or can trigger receptor signaling (by aggregation of multiple receptors). Antibodies can also lead to lysis of cells that express the molecule targeted by the antibody. Target-cell lysis can occur by activation of the complement protein cascade or initiation of antibody-dependent cell-mediated cytotoxicity (ADCC) (Fig. 21–15). The complement cascade is initiated by binding of the Fc domain of immunoglobulin (Ig)G to a complement protein, which then leads to a cascade of cleavage of various complement system proteins. The end result of this cascade is the formation of a protein complex called the membrane attack complex (MAC) on the target cell (in this case, the tumor cell) that leads to pore formation and lysis of the target cell. Other proteins activated by the complement cascade are able to mediate chemoattraction of various immune cells to the site of tumor. ADCC, on the other hand, is initiated upon binding of the Fc region of antibodies by Fc receptors (FcRs). FcRs are expressed on various innate immune cell types, and the activation of FcRs leads to activation of cytotoxic activity against target cells. For example, FcγRIIIA is expressed by natural killer (NK) cells, and binding of this receptor by the Fc region of an IgG molecule activates NK cell-mediated lysis of the target cell. Trastuzumab (Herceptin), which is principally thought to bind and inhibit signaling through the HER-2/neu growth receptor (see Chap. 7, Sec. 7.5.3 and Chap. 20, Sec. 20.3.3), also has been shown to mediate antitumor activity via ADCC (Clynes et al, 2000). Because ADCC is a potentially important mechanism mediating the action of therapeutic antibodies, the most effective antibodies should be engineered or selected to bind well to activating FcRs and to bind poorly to the inhibitory FcγIIB (Nimmerjahn and Ravetch, 2008).
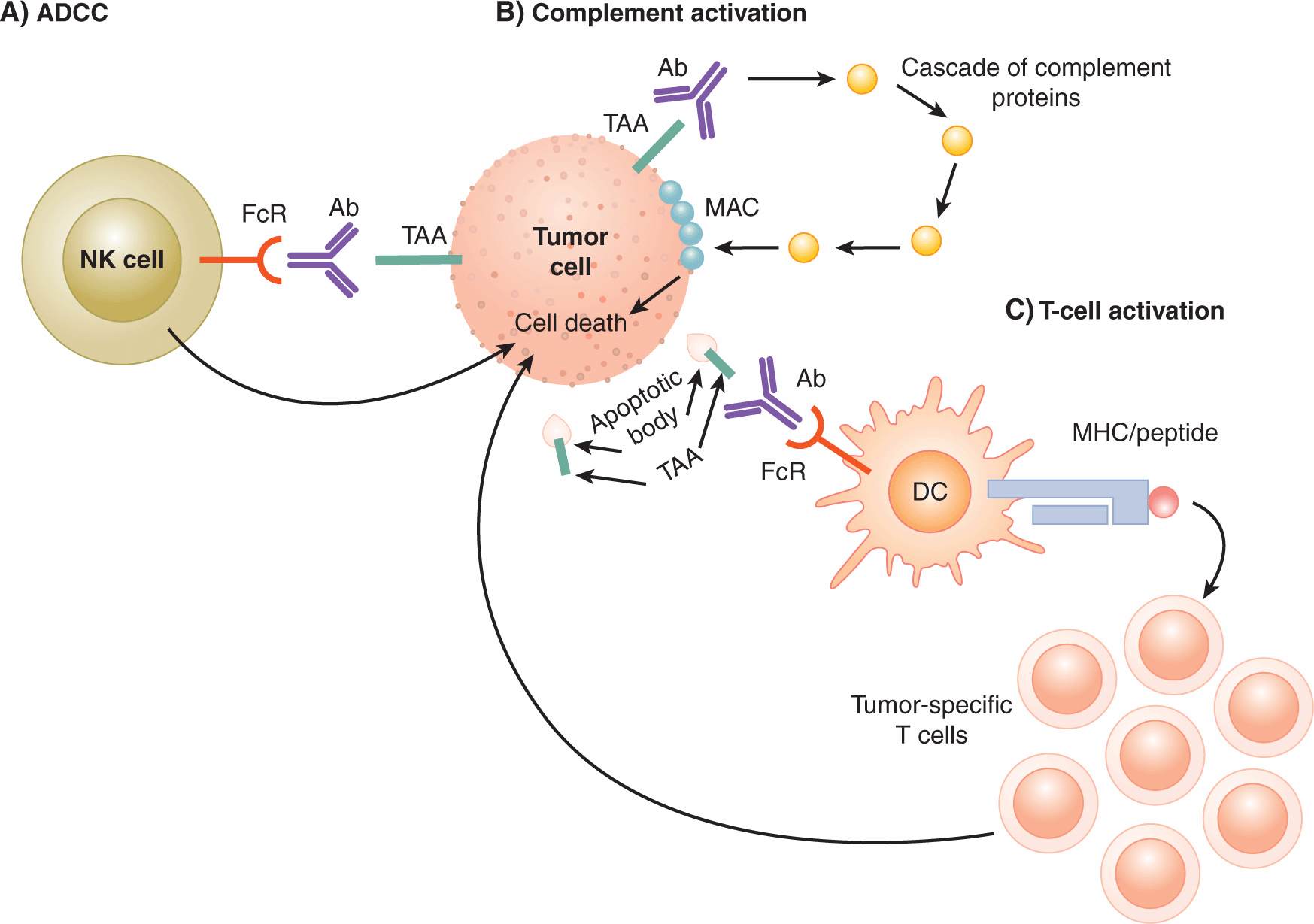
FIGURE 21–15 Tumor-specific antibodies can promote tumor regression by multiple mechanisms. In addition to either blocking or triggering cell-surface receptors, antibodies can lead to immune-mediated elimination of tumor cells. A) ADCC is induced when antibodies bound to tumor antigens (TAA) on the surface of tumor cells are then bound by Fc receptors expressed by natural killer (NK) cells. Triggering of the Fc receptors activates the cytolytic activity of NK cells. B) Complement activation is induced by binding of complement proteins to the Fc domain of antibody/antigen complexes. One of the end results of the complement cascade is the formation of the membrane attack complex (MAC) in the cell membrane of target cells, leading to cell death. C) Antibodies can also bind to tumor antigens present on apoptotic bodies formed when tumor cells undergo apoptosis during the normal cycle of proliferation and cell death. By this route, tumor antigens are taken up by DCs via Fc receptors. Tumor antigens can be processed by the DCs and presented as peptide/MHC complexes to T cells. Tumor-specific T cells can then be activated and mediate antitumor activities including cytotoxic activity against tumor cells.
Antibodies may also lead to enhanced T-cell priming. In the natural course of tumor progression or during antibody-mediated lysis of tumor cells, fragments of dying tumor cells can be recognized and bound by TAA-specific antibodies (see Fig. 21–15). These complexes can then be recognized by FcRs expressed on DCs, internalized, and the TAA-derived peptides presented to tumor-specific T cells via cross-presentation. The relative contribution of each of these different mechanisms for various antibodies is unclear.
21.5.2.4 Modified Antibodies Antibodies can be modified in various ways in order to alter their therapeutic function. For example, bispecific T-cell engager (BiTE) molecules consist of 2 Fv fragments, each with different specificities, linked together with a flexible linker (Wolf et al, 2005). For example, a BiTE has been generated that is specific for CD3 and epithelial cell adhesion molecule (EpCAM). The EpCAM-specific component binds tumor cells expressing this common tumor adhesion molecule and the CD3-specific component functions to recruit T cells to EpCAM-expressing tumor cells. Another example is Blinatumomab, which has been shown to mediate regression of non-Hodgkin B-cell lymphomas (Bargou et al, 2008). This agent is specific for CD19 (expressed on non-Hodgkin lymphoma cells) and CD3 (to recruit T cells to tumor cells).
Three FDA-approved antibodies for cancer have been modified to deliver toxic or radioactive molecules to tumor cells. Gemtuzumab is fused to the toxin ozogamicin, Ibritumomab tiuxetan is conjugated to yttrium-90, and Tositumomab is conjugated to iodine-131 (Bross et al, 2001; Fisher et al, 2005; Witzig et al, 2002).
21.5.3 Nonspecific Immune Modulators
21.5.3.1 Bacillus Calmette-Guérin Bacillus Calmette-Guérin (BCG) is a live-attenuated strain of Mycobacterium bovis and is effective in preventing the recurrence of superficial bladder carcinoma. BCG is administered intravesically, and its mechanism of action is related to its immunostimulatory activity (Patard et al, 1998).
After instillation in the bladder, BCG is processed and presented locally by APCs. These APCs then stimulate CD4 and CD8 T-cell responses. In addition, upregulation of MHC class I and II on urothelial cells is observed. During BCG treatment, levels of various cytokines are found in the urine: IL-1, IL-6, IL-8, and IL-10 early during treatment, and IL-2, TNF-α, and IFN-γ later during treatment. Infiltration of T cells into the bladder wall is also observed after treatment. Overall, BCG instillation leads to a local T-cell response, which appears to have features of a Th1-type response. The mechanism whereby tumor cells are eliminated is unclear. It is possible that myco-bacteria-specific T cells are activated and lyse tumor cells that have been infected with the mycobacteria. It is also possible that the immune stimulation induced by BCG promotes the activation of T cells specific for bladder tumor antigens and that these T cells kill tumor cells.
21.5.3.2 Interferon-α IFN-α is produced by many cell types, including cells of the immune system such as T cells, B cells, NK cells, DCs, and macrophages, and has direct antitumor effects, as well as a role in immunomodulation (Dunn et al, 2006). IFN-α can directly inhibit proliferation of tumor cells, downregulate expression of oncogenes, and induce tumor-suppressor genes. It also possesses antiangiogenic activity.
IFN-α therapy induces responses in more than 90% of patients with hairy cell leukemia, although most patients will relapse within 2 years. IFN-α also shows some activity in patients with other hematological cancers, such as chronic myelogenous leukemia, myeloma, and low-grade non-Hodgkin lymphoma. Although approved by the FDA for patients with melanoma and renal cell carcinoma, the overall clinical response rate in these patients to IFN-α therapy is relatively low. Furthermore, high-dose IFN-α therapy is associated with severe toxicities, including hypotension, vomiting, fever, and diarrhea (Kirkwood et al, 1996; Motzer et al, 2002).
Although the utility of IFN-α therapy is limited, IFN-α is an important enhancer of antitumor immunity. IFN-α induces upregulation of MHC class I and contributes to maturation of DCs. It also is important for promoting CD8 T-cell survival and enhances migration of T cells into tissues. For example, mice treated with an IFN-α receptor-blocking antibody could not reject MCA-induced sarcomas that could be rejected in untreated mice. In addition, IFN-α receptor 1-deficient mice were more susceptible to MCA-induced tumor formation (Dunn et al, 2006). IFN-α may have a therapeutic role in combination with other types of immunotherapy. For example, in a murine model of colon carcinoma, combination therapy using an immunostimulatory antibody (agonistic anti-CD137 mAb) and IFN-α resulted in synergistic antitumor immunity (Dubrot et al, 2011).
21.5.3.3 Interleukin-2 IL-2 is a well-characterized cytokine that acts primarily to stimulate proliferation of T cells and NK cells. High-dose IL-2 therapy is an approved treatment for renal cell carcinoma and melanoma, but the clinical response rate for this therapy is only in the range of 15% for both cancer types (Atkins et al, 1999; Fyfe et al, 1995). High-dose IL-2 therapy is also associated with substantial toxicity, including cardiac arrhythmias, capillary leak syndrome leading to hypotension, central nervous system toxicity leading to confusion and gastrointestinal toxicity such as diarrhea (Rosenberg et al, 1989). Although IL-2 promotes effector T-cell responses and can function to reverse anergy of effector T cells, IL-2 may not represent an ideal agent to promote immunity because it also promotes the expansion of Tregs.
21.5.3.4 Imiquimod Imiquimod is a synthetic compound that triggers the TLR7 molecule. TLRs are a family of molecules that are stimulated by conserved molecular motifs that are present on various microorganisms (Akira et al, 2006; see also Sec. 21.2.2). For example, the natural ligand for TLR7 is single-stranded RNA, which is present in some viruses. The TLRs are expressed by many cell types, including cells of the innate immune system such as macrophages and DCs. Interactions between TLRs and corresponding ligands lead to activation of innate immune cells, which promote elimination of the microorganism through various mechanisms, including production of interferons and proinflammatory cytokines.
Because TLRs activate APCs such as DCs, they can enhance the ability of DCs to stimulate antitumor T-cell responses. Various TLR ligands are under investigation in animal models and early clinical trials for cancer immunotherapy. These agents are often used in combination with vaccination strategies to enhance T-cell responses induced by vaccination.
Imiquimod is approved for use in the treatment of superficial basal cell carcinoma, and for treatment of genital warts and actinic keratosis. Treatment is generally effective for superficial basal cell carcinoma, with complete clearance rates of approximately 75% or higher (compared to a few percent in placebo-treated groups). Imiquimod has been shown to induce proinflammatory mediators such as IFN-α, TNF-α, and IL-6 by various innate immune cell types, including macrophages and a specialized subset of DCs called plasmacytoid DCs. Imiquimod also upregulates costimulatory molecules and chemokine receptors on plasmacytoid DCs, thus enhancing their ability to activate T cells and home to sites of T-cell activation, respectively. Histological studies have shown an increase in infiltration of tumor lesions with T cells and DCs following Imiquimod treatment, further supporting its immunological mechanism of action.
21.5.4 Other Approaches to Immunotherapy
21.5.4.1 Dendritic Cell Vaccines Mature DCs are highly efficient APCs and therefore are central players in the induction of T-cell responses. Therefore, adoptive cell therapy using DCs that present TAAs to induce antitumor T cells in vivo is of major interest (Tacken et al, 2007). In general, these DCs are obtained by in vitro differentiation of bone marrow progenitor cells (for mice) or peripheral blood monocytes (for humans). Granulocyte macrophage-colony stimulating factor (GM-CSF) and IL-4 are commonly used to stimulate differentiation. Figure 21–16 outlines the generation of DCs for infusion. Some of the methods for loading DCs with TAAs include loading peptides derived from TAAs into surface MHC molecules, incubating DCs with whole TAA proteins, and transfecting or transducing DCs with DNA encoding TAAs. TAA-loaded DCs may also be exposed to a variety of maturation stimuli in order to enhance their immunogenicity. These stimuli include agonistic anti-CD40 antibody, various TLR ligands (eg, CpG oligodeoxynucleotides, poly I:C, LPS), and cytokine cocktails (eg, a cocktail of IL-1β, IL-6, TNF, and prostaglandin E2).
FIGURE 21–16 Generating DCs for therapy. Common methods for generating DCs for vaccination are shown. A) For mouse studies, DC precursors are obtained from bone marrow cells and differentiated into immature DCs in vitro using GM-CSF. (B) For human studies, monocytes are obtained from peripheral blood mononuclear cells by a variety of methods, including elutriation, plastic adherence, or isolation of CD14+ cells. Monocytes are then differentiated into immature DCs in vitro using GM-CSF and IL-4. For both mouse and human studies, immature DCs are manipulated to present tumor-associated antigens. This can be done by a variety of approaches, including exposing them to peptides or RNA, or fusing the DCs with tumor cells. DC maturation is induced by stimuli such as TLR ligands (eg, LPS or CpG oligodeoxynucleotides) or agonistic anti-CD40 antibody. Mature DCs presenting TAAs are then ready for infusion.
Clinical trials based on DC vaccination have demonstrated that this approach is associated with minimal toxicity, and there is some evidence for clinical effectiveness (Tacken et al, 2007; Palucka et al, 2012).
21.5.4.2 Sipuleucel-T Sipuleucel-T was the first autologous cellular therapy for cancer to receive FDA approval. The therapeutic product is produced by leukapheresis from a patient with prostatic cancer. The leukapheresis product is transported to a central facility where the cells are processed and incubated with a fusion protein of a prostate TAA (prostatic acid phosphatase [PAP]) and the cytokine GM-CSF (Small et al, 2000). The cellular product is then reinfused into the same patient. The premise of this therapy is APCs, such as DCs, will take up the fusion protein. The PAP will be processed and presented by MHC molecules on the surface of APCs and will thus be able to stimulate PAP-specific T cells to mediate antitumor activity. The functions of GM-CSF include promoting the differentiation of DCs. Presentation of a prostate TAA by efficient APCs such as DCs should activate endogenous prostate-specific T cells upon reinfusion. However, it remains unclear how the other cell types present in the leukapheresis product may contribute to the effectiveness of the treatment. In vitro biological potency tests as well as clinical trial data indicate that the activity can be attributed to the cell fraction expressing CD54, which is a cell adhesion molecule expressed on a variety of cell types, including APCs. An integrated analysis of 2 Phase III trials showed a survival benefit for advanced prostate cancer patients treated with sipuleucel-T compared to placebo (Higano et al, 2009), with a 23.2-month median survival in the Sipuleucel-T arms and an 18.9-month median survival in the placebo arms. Common adverse events included chills, pyrexia, and headache, and were mostly low grade. An additional double-blind, multicenter Phase III trial showed similar results. In this trial, 512 men with metastatic castration-resistant prostate cancer were randomized at a 2:1 ratio to receive Sipuleucel-T or placebo (Kantoff et al, 2010). Interestingly, the time to objective disease progression was similar in both arms, but the relative risk of death was reduced by 22% in the Sipuleucel-T arm compared to the placebo arm, leading to a 4-month improvement in median survival. However, this trial has been criticized because the leukapheresis procedure was used in control patients (but leukocytes were not re-infused into all of the control patients), and loss of white cells might be harmful, particularly to older adults, leading (in part) to a difference in survival because of poorer outcome in controls (Huber et al, 2012).
21.5.4.3 CTLA-4 Blockade CTLA-4 is expressed on activated T cells and also Treg cells. Initial experiments in animal models demonstrated that blockade of CTLA-4 induces tumor regression. Two therapeutic anti–CTLA-4 blocking antibodies have been developed. One is a fully human IgG2 antibody (Tremelimumab) (Ribas, 2008), the other is a fully human IgG1 antibody (Ipilimumab) (Weber, 2008). These agents have been tested mainly in patients with metastatic melanoma.
The clinical activity of these agents, when assessed initially using objective response (ie, tumor shrinkage) to evaluate clinical responses, appeared to be relatively low. However, this type of immunomodulatory agent may induce unconventional response patterns, such as tumor progression that precedes regression, or mixed responses of different lesions but with overall decreases in tumor burden (Wolchok et al, 2009). Indeed, a Phase III randomized trial in patients with unresectable Stage III or IV melanoma demonstrated that patients that received Ipilimumab had a significantly longer median overall survival (10.0 months) compared with patients not receiving anti–CTLA-4 blockade (6.4 months) (Hodi et al, 2010). Because anti–CTLA-4 blockade is not an antigen-targeted approach, it is associated with some grade 3 or 4 autoimmune toxicities, the most common being colitis. In 2010, Ipilimumab received FDA approval for treatment of patients with unresectable or metastatic melanoma.
Although the negative regulatory role of CTLA-4 signaling is well established, the mechanism(s) whereby anti–CTLA-4 blockade induces antitumor T-cell immunity remains under investigation. There remains some debate whether the predominant target of anti–CTLA-4 blockade is to “release the brakes” on effector T cells or to inhibit the suppressive activity of Tregs. CTLA-4 blockade is thought to enhance effector T cells by the following mechanisms: Anti–CTLA-4 antibodies may prevent the interaction of CD80 and CD86 with CTLA-4, thereby allowing for prolonged interaction of CD80 and CD86 with the positive costimulatory receptor CD28. In addition, CTLA-4 blockade may inhibit the CTLA-4–mediated negative intracellular signals that shut down effector T cells. There is also strong evidence that CTLA-4 suppresses effector T cells in a non–cell-autonomous fashion, that is, by inhibiting the suppressive activity of Tregs (Bachmann et al, 1999; Read et al, 2000). Evidence suggests that the effects of CTLA-4 on both effector T cells and Tregs contribute to its ability to enhance antitumor immune responses (Peggs et al, 2009).
21.5.4.4 Adoptive T-cell Therapy Infusion of T cells that can recognize and destroy tumor cells is a major area of interest for immunotherapy. That transferred T cells can mediate potent antitumor effects has been conclusively demonstrated by donor lymphocyte infusions (DLIs), which are the standard treatment for patients with relapsed leukemia following allogeneic bone marrow transplantation. For therapy using DLI, lymphocytes from allogeneic bone marrow transplantation donors are stored in a sample bank. If the leukemia patient relapses following a bone marrow transplantation, then the lymphocytes from the same donor are infused into the patient. These lymphocytes mediate regression of the relapsed leukemia (a so-named graft-versus-leukemia effect). For patients with relapsed chronic myelogenous leukemia, 70% of patients treated with DLI experience complete remission (Kolb et al, 2004). This approach demonstrates that T cells have the potential to mediate antitumor activity.
Adoptive T-cell therapy is based on ex vivo expansion and manipulation of patient-derived tumor specific T cells followed by reinfusion in an autologous manner. General approaches to generate T cells for adoptive cell therapy are depicted in Figure 21–17 and described below.
FIGURE 21–17 Generating T cell clones or tumor-infiltrating lymphocytes (TILs) for adoptive cell therapy. A) To generate T-cell clones, peripheral blood mononuclear cells are isolated and the bulk T-cell population is stimulated with DCs that have been exposed to peptides derived from tumor-associated antigens. Through successive rounds of stimulation and expansion, tumor-specific T cells are selectively expanded. These T cells are cultured under “limiting dilution” conditions to generate cultures originating from 1 parent cell (a “clone”) consisting of T cells with the same T-cell receptor. B) To generate TILs, tumor tissue is dissociated and the bulk population of TILs is expanded in vitro using a T-cell growth factor (eg, IL-2). In theory, the resulting T-cell population is enriched for tumor-specific T cells.
21.5.4.4.1 Adoptive Cell Therapy with T-cell Clones One source of T cells for adoptive cell therapy is expanded T-cell clones from the peripheral blood of cancer patients. This approach is advantageous in that the peptide specificity of the transferred T cells is well defined, and peripheral blood T cells have not been subjected to immunosuppressive factors present within the tumor microenvironment and therefore may be more responsive. Potential drawbacks for this approach include the possibility that these T cells may not home to the tumor site. It is also possible that tumor cells do not express the peptide recognized by the T-cell clones and will escape detection by the T cells.
The general approach to generating T-cell clones for therapy involves stimulation of bulk peripheral blood T cells with peptides derived from TAAs of interest. The most well-studied TAA-derived peptides are those that bind the HLA-A*0201 MHC molecule, and therefore trials of adoptive cell therapy using clones are generally limited to HLA-A*0201–positive patients. Peptide-stimulated T cells are then cultured under limiting dilution conditions in order to expand a population of T cells derived from a single clone. All T cells expanded from a single T cell will express identical TCRs and therefore can be considered clones. CD8+ T-cell clones and, more recently, CD4+ T-cell clones have been used in early phase clinical trials that have demonstrated this approach is associated with low toxicity (Yee, 2010).
21.5.4.4.2 Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes Many solid tumors are infiltrated with T cells. Because, under normal circumstances, T cells do not infiltrate tissues, the presence of tumor-infiltrating T cells (TILs) suggests that an antitumor response is occurring and the T-cell infiltrate is likely enriched for those that can recognize TAAs. Therefore, TILs represent a source of tumor-specific T cells for use in therapy. TILs can be obtained following dissociation of tumor tissue and expansion of TILs can be undertaken ex vivo in the presence of various T-cell growth factors such as IL-2. Adoptive transfer of bulk populations of TILs has been performed in trials for various cancers, including melanoma, ovarian cancer, renal cell carcinoma, and non–small cell lung cancer. Collectively, these trials demonstrate that adoptive transfer of TILs is associated with minimal toxicity and some of these studies provide evidence that TILs are clinically active.
High clinical response rates have been observed when patients with metastatic melanoma were treated with TIL-based protocols in a particular series of trials. In these protocols, patients were given nonmyeloablative lymphodepleting chemotherapy (cyclophosphamide and fludarabine) immediately prior to infusion of TILs (1010 to 1011 cells) and high-dose IL-2 therapy. Using this protocol, the objective clinical response rate by RECIST (Response Evaluation Criteria in Solid Tumors) criteria was approximately 50% (21/43 patients) (Dudley et al, 2002, 2005). When total body irradiation was added to the treatment protocol, a trend to higher clinical response rates with increasing intensity of lymphodepletion was observed (Dudley et al, 2008). Therefore the combination of TIL transfer with other therapeutic interventions has the potential to improve disease outcomes, although this has not yet been tested in Phase III trials.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


