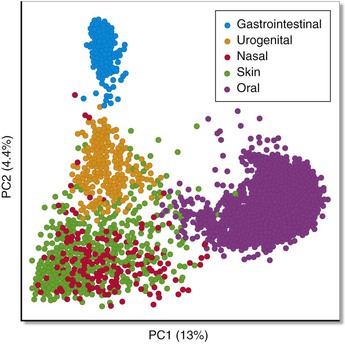
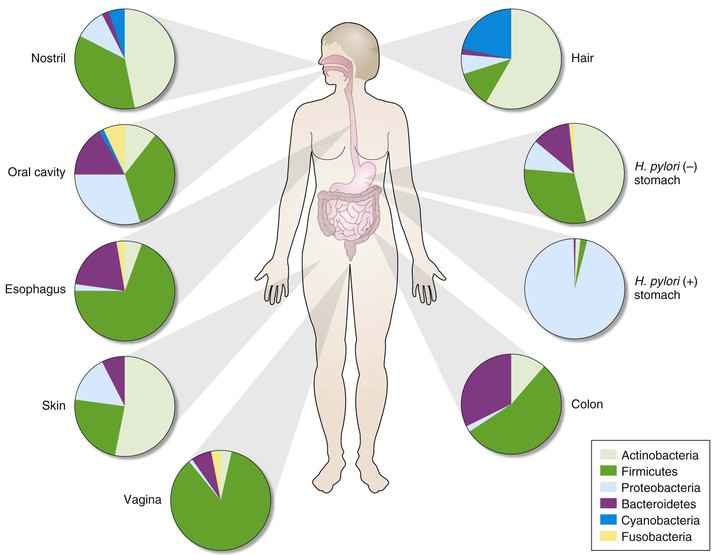
Kjersti Aagaard, Ruth Ann Luna, James Versalovic The human microbiota can be defined as all microorganisms (≈90 or so trillion bacteria, Archaea, microeukaryotes, and viruses) residing in the human body; the human microbiome consists of the genes and gene products (RNA, proteins, metabolites) produced by resident microbial communities. The advent of high-throughput DNA/RNA sequencing technologies and computational methodologies has enabled scientists to systematically catalog the global set of microorganisms—cultured and uncultured—in a heretofore unparalleled manner. Different body habitats contain microbial communities and microbiomes that differ by microbial composition and function (metabolic modules and pathways). As a result, each body habitat is composed of characteristic bacterial species and other microbial taxa that are adapted to each body site. Differences in microbial composition yield differences in metabolic capacity and aggregate function of the human microbiome. Traditional notions have been challenged, such as the ideas first put forth in Koch’s postulates, whereby microbes were viewed as pathogens and as sole etiologic agents of infectious diseases. Such a “foe” view neglects our earliest sightings of oral and fecal microbes with Anton van Leeuwenhoek’s microscopes, where it was observed that animalcules (microorganisms) reside in a symbiotic and likely mutually beneficial relationship with the host. We now appreciate that the microbial genome exceeds the human genome by at least 250-fold, and the cellular count of resident microbiota exceeds the human cell count by greater than 10-fold.1 Our concepts regarding the relative abundance and ubiquity of diverse human pathogens are growing more profoundly with advances in the science of the human microbiome. Abundance refers to the relative quantity of microbes within each individual or body site, whereas ubiquity refers to the presence of the same microbes in different individuals. The Human Microbiome Project (HMP) documented the striking absence of canonical pathogens in healthy adults at 18 body sites.2 Notable exceptions were the well-known pathogens Staphylococcus aureus and Escherichia coli. As an example, E. coli DNA was detected in 15% of individuals at 0.5% abundance and was detectable at any level in 61% of healthy adults. Canonical pathogens as defined by the National Institute of Allergy and Infectious Diseases2 are generally absent from the human microbiome in healthy individuals, but opportunistic pathogens are widely distributed in healthy adults. A total of 59 opportunistic pathogens in the PathoSystems Resource Integration Center (PATRIC) database were detected in 242 healthy adults, and these species were shared in colonized individuals across multiple body sites. This finding contrasts with the relative habitat specificity of commensal species that lack evidence of pathogenicity. In summary, although canonical pathogens are rare in healthy individuals, opportunistic pathogens are relatively common in healthy individuals and explain why immunosuppression often results in opportunistic infections. Canonical pathogens, by contrast, must be transmitted to healthy individuals from other humans, animals, or the environment. Opportunistic pathogens may arise from within the indigenous microbiome, in addition to possible transmission from outside sources. The HMP (funded by the U.S. National Institutes of Health) and Metagenomics of the Human Intestinal Tract (MetaHIT; funded by the European Commission) initiatives established the first microbial gene catalogs of the human adult microbiota, with the HMP effort spanning 15 body site niches in men and 18 in women.1–4 Each primary body habitat in the healthy human microbiome contains a distinctive microbial community, when evaluated by bacterial composition.2,3,5 With the establishment of these first reliable estimates of the structure, function, and diversity of the human “healthy” microbiome, it is readily apparent that there is body site (niche) specificity (Table 2-1, Fig. 2-1). The HMP reported that although no bacterial taxa were universally present among all body habitats and individuals, the relative distribution of several metabolic modules and pathways was surprisingly similar, with a greater degree of similarity observed within ethnic and racial groups.2 These carriage patterns were functionally relevant, and genomic variation in microbial strains (gains, losses, and polymorphisms) underscored the extent of interindividual variation in the human microbiome. Taxonomic profiling associating both clades and metabolism with host covariates (namely, adult age, sex, body mass index [BMI], blood pressure, race, and ethnicity) demonstrated that most microbial variation is not well-explained by examined clinical covariates other than race/ethnicity.4 TABLE 2-1 Explanation of Key Terms As a result, our rapidly evolving view of the human ecosystem augments the traditional view of a single pathogen being responsive for disease onset. Even if a single microbe is the etiologic agent of infection, the pathogenesis and pathophysiology of infection can be viewed within the context of the microbiome and human biology. We now appreciate that our human microbiome is a complex ecosystem, with distinct biologic niches. The resultant perspective for human health and disease shifts the focus to the global balance of our microbiota rather than the appearance of a specific infectious agent. As a result, a clear understanding of the role of microbial community structure in the host can facilitate a deeper understanding of infectious diseases and susceptibility to infections (see Table 2-1). We are realizing the translational fruits of a broadened understanding of the human microbiome as metagenomic medicine makes strides in restoring health in highly morbid conditions (e.g., recurrent Clostridium difficile colitis).6 The period of establishment of the human microbiome in newborn infants is highly debated. Several theories have been proposed, such as acquisition from exposure to the maternal vaginal microbiome, intestinal microbiome, breast milk microbiome, and skin-to-skin contact7; however, these findings are not exclusive. In murine models, the newborn gut is relatively sterile before birth and soon after delivery is exposed to both the maternal and environmental microbiota.8 Symbiotic and commensal microbiota in the human host may prevent colonization by harmful bacteria and provide immune resistance as demonstrated by germ-free mouse experiments. The mode of delivery has been associated with differences in composition of the newborn microbiome.9–13 As the infant passes through the birth canal, the maternal vaginal microbiome is likely in contact with the infant’s skin, mouth, and respiratory tract, and this exposure may affect the composition of the offspring’s skin, oral, intestinal, and nasal microbiomes. In one of the often-cited studies regarding mode of delivery and the impact on the human microbiome, investigators examined neonatal microbial community structure in instances of cesarean delivery in comparison with vaginal birth from a small cohort of Venezuelan women employing 16S-based metagenomics.10 They observed that the vaginally delivered infants harbored Lactobacillus, Prevotella, or Sneathia spp. (commonly found in the vagina), whereas cesarean-delivered infants acquired a microbiome that was dominated by Staphylococcus, Corynebacterium, and Propionibacterium spp. (resembling the skin microbiota of their mothers and others in the local environment).10 Interestingly, although infants delivered by cesarean were more akin in their microbiome profile to human skin microbiome communities, the mother’s skin microbiome did not bear a closer resemblance to her respective infant’s microbiome when compared with other babies born via the same mode of delivery or vaginally delivered maternal-infant pairs.10 This finding suggests that vertical transmission may occur with vaginal birth, but horizontal transmission of microbes from other humans and the environment contributes to diversification of the skin microbiome after cesarean birth. In terms of the longitudinal establishment of the human microbiome, the microbiomes in vaginally versus cesarean-delivered infants yielded a modest but significant difference at up to 6 months of age and appreciable differences years later.9,12,14 Neonates delivered by cesarean section yield a characteristic absence of Bifidobacterium spp., whereas those infants delivered vaginally have a predominance of Bifidobacterium longum and Bifidobacterium catenulatum.9,13 The composition of the newborn microbiome and its link to mode of delivery is confounded by the fact that mothers undergoing cesarean section routinely receive antibiotics.12 Maternal factors, such as the indication for cesarean, use of preincision versus perioperative antibiotics, consumption of probiotics, maternal BMI, and gestational diabetes status, are understudied likely modifiers.8–10,14 These early observations regarding mode of delivery and its impact on the establishment of the microbiome are not yet reported in broad population-based cohorts, and potential underlying co-associations and comorbidities deserve greater attention in future studies. In this chapter we describe the current state of knowledge of the human microbiome and key features of human-associated microbial communities in each primary body habitat. We render brief discussions regarding known determinants of the microbial structure of these niches and presumptive associations with several disease states (as examples). Because of the paucity of data pertaining to a definitive role of variance in the microbiome as the source of disease, we limit the entirety of our discussion in this chapter to co-occurrence and associations, rather than presumptive causation. The oral microbiome is diverse and abundant. Although a significant amount of research has focused on the gut microbiome with respect to health and disease, there is a substantial body of work regarding the oral microbiome. The HMP demonstrated exquisite niche specificity within the oral microbiome, with distinct communities observed at the level of taxa and gene carriage patterns (Fig. 2-2). To put this into perspective, 1 cc (mL) of human saliva in a healthy adult contains approximately 100 million cells, which is discrete from the community of the surrounding oral microbiome. Several studies2,3,15–18 have documented the unanticipated robustness of the oral microbiome. Microarray, early pyrosequencing, and culture methodologies estimated approximately 700 oral microbial phylotypes. However, dental plaque sampling pooled from 98 healthy adults was estimated to represent 22 phyla comprising 3621 and 6888 species-level phylotypes in the saliva and plaque, respectively.15 The HMP estimated nearly 70 distinct genera in the same human specimen types.2 The most abundant bacterial genera in healthy adults include Actinomyces, Bacteroides, Prevotella, Streptococcus, Fusobacterium, Leptotrichia, Corynebacterium, Veillonella, Rothia, Capnocytophaga, Selenomonas, and Treponema, as well as the TM7 lineage. In addition to the bacterial kingdom, Methanobrevibacter spp. from the Archaea kingdom was also identified in the oral microbiome. Interindividual variation in the microbiome is richly observed in the oral niche. For example, Streptococcus spp. dominate the oropharynx,2 with exquisite strain-level genomic variation within microbial species and enriched for host-specific structural variants around genomic islands. Although these differences are also observed in the gut and skin (leading to issues such as methicillin-resistant S. aureus), the oral microbiome is unique in its maintenance of closely adjacent subsites within the niche. The tonsil microbiome can be distinguished from the tongue, and the tongue from the palate. These differences are evident despite spatial proximity and constant contact between these sites. With maintenance of niche and subsite specificity in mind, it is not surprising that long-standing associations have been documented between oral health and disease manifestations in distal body sites. For example, periodontal disease is the most common infectious disease affecting the teeth. Left untreated or ineffectively treated, periodontitis is a known independent predictor of, and comorbid contributor to, preterm birth, cardiovascular disease, pulmonary disorders, diabetes, and obesity.19 Additional strong correlations between the qualitative composition of the oral microbiota as a whole have been made with different disease states. The generation of the dental plaque biofilm that we experience daily has been well characterized.15–19 A succession of early and late colonizing species, dominated early by Streptococcus spp., coat the dentin surface of the tooth. Once this early biofilm establishes, a series of highly coevolved oral bacterial and host interactions occur to likely layer bacteria on bacteria, which ultimately generates a large and diverse microbiota load on the tooth surface with a generally healthy periodontium (see Table 2-1). Periodontitis may be considered akin to bacterial vaginosis (BV) or inflammatory bowel disease (IBD) because it is not the singular absence or presence of a given species or subgenus that drives the oral gum inflammation. Rather, the complexity of the subgingival microbiota and biofilm establishment promote a model of a microbial community-associated disease. In one recent study using deep sequencing, periodontitis was associated with a shift to populations enriched with gram-negative genera such as Catonella, Haemophilus, and Tannerella.18 The microbiome-minded clinician scientist will consider the stability of oral microbiome composition as a hallmark of human health. One exception is infection by Aggregatibacter actinomycetemcomitans because this gram-negative rod appears to cause a highly aggressive periodontitis (localized aggressive periodontitis) in Africans with strong host tropism.19 Other investigators have used whole genome shotgun sequencing in smaller-scale studies to suggest a potential role for uncultured bacteria of the TM7 lineage in other forms of periodontitis, but broader health and disease associations have yet to be tested in a population-based cohort. As the integument (including skin, hair, and nails) that comprises the main body surface in constant contact with the outside environment, the human skin consists of diverse sets of local habitats and niches for the human microbiome. The human skin comprises various ecosystems that differ markedly by relative differences in temperature, humidity, and glandular distribution. The human skin microbiome and the nature of the local environment can vary greatly depending on anatomic location. One report described bacterial compositional differences in 20 different sites on the human skin.20 Recent studies have demonstrated that the skin microbiome differs among healthy individuals more than any other site.2,3 Bacterial communities are composed largely of transient (superficial), autochthonous (deep surface), and adherent microbes lying adjacent to the epidermis, but they have also been detected in the subepidermal compartment including the dermis and dermal adipose tissue.21 Different factors contribute to variation of the human skin microbiome. These factors include host physiology (sex, age, site); environment (local climate, geographic location); immune system; host genotype; lifestyle (occupation, hygiene); and pathobiology (skin and systemic diseases).22 Different regions of the human skin contain characteristic distributions of different types of glands. These glands produce oily substances such as sebum and other lipid, carbohydrate, and proteinaceous components that may serve as nutrients for the microbiome, as well as inhibitors to particular classes of microbes. For example, sebaceous gland–rich regions include the head, shoulders, upper arms, and upper torso.20,23 Eccrine glands are most abundant in the crown of the head, under the arms, and the palmar surfaces of the hands. Apocrine glands are enriched around the eyes and ears, nipples, and genital regions. Relative humidity is another key factor affecting microbial composition of the skin. Areas rich in sebaceous glands are enriched for Propionibacterium spp., whereas moist and dry skin areas are enriched for Corynebacterium spp. and β-Proteobacteria, respectively.20 The HMP provided the most comprehensive survey of the human skin microbiome in terms of the number of different male and female adult individuals.2 A total of 242 individuals were fully analyzed at three body sites (anterior nares, antecubital fossa, retroauricular crease). This study confirmed that the skin microbiome is distinct from that of other body sites and it is characterized by an intermediate degree of alpha diversity and richness per specimen. The phyla Actinobacteria, Firmicutes, and Verrucomicrobia were the dominant groups in the human skin,24 in contrast with the predominance of Bacteroidetes, Firmicutes, and Proteobacteria in the human gut. Therefore, even at the level of phyla composed of hundreds of different bacterial species, stark differences are evident in the skin compared to other body sites.25 Results depend on technical considerations and different specimen types such as skin swabs, blade scrapings, and skin biopsy specimens.26 The most commonly accepted specimen type currently for human microbiome studies is skin swabs, partly due to the diminished amount of contaminating human microbial DNA in more superficial samples.3 Adequate amounts of microbial DNA have been generated for 16S rRNA gene sequencing and whole metagenome sequencing using swab specimens, although specific microbes may be present only in deeper specimens or human tissue. For evaluation of dermatologic infections, deeper surface and tissue sampling may be necessary in order to detect infectious agents. Age is an important factor as evidenced by shifts in bacterial communities that occur during the sexual maturation process.27 Corynebacteriaceae and Propionibacteriaceae predominated in Tanner 5 individuals compared with Tanner 1 children. Specific groups of microbes may be conserved in the skin of healthy individuals, whereas interindividual variation may account for differences in relative abundances of microbes and differences in disease susceptibilities.22 Different proportions of Propionibacterium spp. and β-Proteobacteria, for example, may be present on the backs or arms, respectively, in different individuals, but these bacterial groups are present in the majority of healthy individuals at these body sites. Representative bacterial genera in the human skin across sites include Corynebacterium, Eubacterium, Propionibacterium, Staphylococcus, and Streptococcus,24 as well as the fungal Malassezia spp.28 In the nares, Corynebacterium is the most common bacterial genus,22 and persistent S. aureus colonization was found in the nares in 24% of healthy human subjects.29 The nasal microbiota contains different proportions of staphylococci with some individuals containing mostly S. aureus and some individuals carrying mostly Staphylococcus epidermidis. The genus Malassezia is the predominant fungal genus of the human skin at multiple body sites, including the head, torso, arms, and legs,24 except for sites on the foot.28 Persistent effects of antifungal agents on skin fungi or “mycome” were observed in this study.28 Relative distributions of commensal bacteria and opportunistic pathogens may help explain different patterns of cutaneous infections and systemic penetration of pathogens from skin surfaces. Hospitalized individuals with S. aureus–predominant nasal microbiomes were also more likely to contain methicillin-resistant S. aureus (MRSA) associated with their hospitalizations.30 In cases of atopic dermatitis, staphylococci including S. aureus and S. epidermidis populations appear to “bloom” and contribute to disease flares and relapse at specific skin sites.31 Bacterial composition of the human skin may be useful for forensic applications. One study described the utility of skin fingertip microbiome patterns for tracking the use of keyboards and perhaps other devices by specific individuals.32 This study highlights the individualized nature of the skin microbiome at specific sites and the fact that interindividual differences in bacterial composition may help explain relative disease susceptibilities.
The Human Microbiome of Local Body Sites and Their Unique Biology
Defining the Human Microbiome
Human Microbiome as a Complex Ecosystem Composed of Multiple Body Site Habitats and Niches
TERM
DEFINITION
Biofilm
A physically (and often temporally) structured aggregate of microorganisms, often containing multiple taxa, and often adhered to each other and/or to a defined substrate
Community structure
Used most commonly to refer to the taxonomic composition of a microbial community; can also refer to the spatiotemporal distribution of taxa
Diversity
A measure of the taxonomic distribution within a community, in terms of either distinct taxa or their evolutionary/phylogenetic distance
Germ free
A host animal containing no microorganisms
Gnotobiotic
A host animal containing a defined set of microorganisms, either synthetically implanted or transferred from another host; often used to refer to model organisms with humanized microbiota
Metagenome
The total genomic DNA of all organisms within a community
Metabonome
The total metabolite pool (and possibly fluxes) of a community
Metaproteome
The total proteome of all organisms within a community
Metatranscriptome
The total transcribed RNA pool of all organisms within a community
Microbiome
The total microbial community, genes, and biomolecules within a defined environment
Microbiota
The total collection of microorganisms within a community, typically used in reference to an animal host
Probiotic
A viable microorganism consumed by the host with direct or indirect health benefits
16S rRNA
The transcript of the 16S ribosomal subunit gene, the smaller RNA component of the prokaryotic ribosome, used as the most common taxonomic marker for microbial communities
Alpha diversity
Within-sample taxonomic diversity
Beta diversity
Between-sample taxonomic diversity
Functional metagenomics
Computational or experimental analysis of a microbial community with respect to the biochemical and other biomolecular activities encoded by its composite metagenome
OTU
Abbreviation for operational taxonomic unit, a cluster of organisms similar at the sequence level beyond a specific threshold (e.g., 95%) used in place of species or genus
Phylochip
A microarray containing taxonomic (and sometimes functional) marker sequences
Oral Microbiome
Associations Between Oral Microbiota and Disease States
Skin and Nasopharynx
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


