(1)
Mayo Clinic, Jacksonville, Florida, USA
2.1 Introduction
Throughout the world diabetic retinopathy (DR) has emerged as a major cause of permanent loss of vision among people over the age of 20 years. Retinopathy has generally been considered a vasculopathy that results from breakdown of the blood-retinal barrier and closure of retinal capillaries. Recent evidence, however, suggests that DR begins as a neuro-retinopathy with vascular changes occurring later during the disease.
Knowledge of retinal anatomy and pathophysiology are important to surgeons so that diagnoses and disease characterizations can be made accurately. Additionally, a detailed understanding of vitreoretinal anatomy enables surgeons to correctly deliver intravitreal depot injections of pharmaceutical agents and perform complex vitreoretinal surgery.
The biochemical abnormalities responsible for the development of diabetic retinopathy may not be important during the day-to-day practice of ophthalmology, but these abnormalities form the base of knowledge from which drugs have been developed to treat DR. Biochemical pathways that are pertinent to the development of diabetic retinopathy are complex and highly interrelated. Recognizing pivotal molecules such as vascular endothelial growth factor (VEGF) from within the biochemical milieu is an extraordinarily difficult process but one that is critical to drug development.
This chapter will cover many of the important highlights of retinal biochemistry in the setting diabetes, though a deep analysis of all affected pathways is beyond the scope of a single book chapter. The chapter begins with a discussion of retinal anatomy, then transitions to the microscopic composition of the blood-retinal barrier, and the biochemical abnormalities found in eyes affected by diabetes mellitus (DM). Finally, a brief discussion of the funduscopic characteristics of diabetic retinopathy will be provided.
2.2 The Retina
2.2.1 History
The retina was first described by Herophilus of Chalcedon (c. 300 B.C.) and it was eventually named by Rufus of Ephesus (c. 110 A.D.). To early anatomists the retina appeared as a net that surrounded and supported the vitreous. Galen noted structural similarities between the retina and the brain, but he did not understand the retina’s function. Kepler was the first to suggest that the retina served as the eye’s primary photoreceptor tissue. Treviranus (1835) performed the first detailed microscopic analysis of retinal structure by employing alcohol fixation. Only with the use of electron microscopy, trypsin digest, clinical fluorescein angiography, and optical coherence tomography were scientists able to understand the retina’s cellular connections, ultrastructure, and retinal vasculature, and ultimately correlate anatomic and clinical findings [169].
2.2.2 Anatomy
The retina is a translucent tissue that lines the posterior two-thirds of the eye from the macula posteriorly to the ora serrata anteriorly [87] (Fig. 2.1). The retina is contiguous with the optic disc since axons of the retinal nerve fiber form the optic nerve as they leave the eye. Anteriorly, the retina attaches to the nonpigmented epithelium of the pars plana at the ora serrata. Interdigitations between the photoreceptor outer segments and the cells of the retina pigment epithelium weakly attach the outer retina to the wall of the eye. The retina is internally attached to the vitreous at the optic nerve, macula, retinal vessels, and vitreous base.
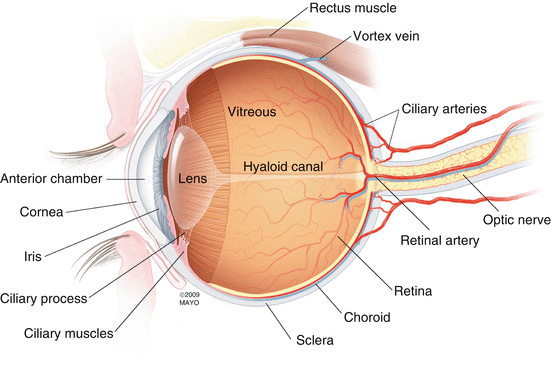
Fig. 2.1
This drawing details the important anatomic and circulatory structures of the eye and supporting tissues. The retina lines the inner, posterior two-thirds of the globe from the optic nerve to the ora serrata (just beyond the termination of the retinal arterioles). The vitreous is the most voluminous (4 ml) tissue within the eye
The central 5–6 mm of the retina, referred to as the retina centralis, has more than one layer of ganglion cells (Fig. 2.2). The central 1.5 mm of the retina is bounded by the termination of the retinal capillary circulation and is called the macula lutea because of the high concentration of xanthophyll in the ganglion and bipolar cell layers [218]. The 0.35 mm depression within the central macula, called the fovea by clinicians and the foveola by anatomists, has only cones in the photoreceptor layer. The center of the foveal depression, the clivus, is situated 3.4 mm temporal and 0.8 mm inferior to the center of the optic disc.
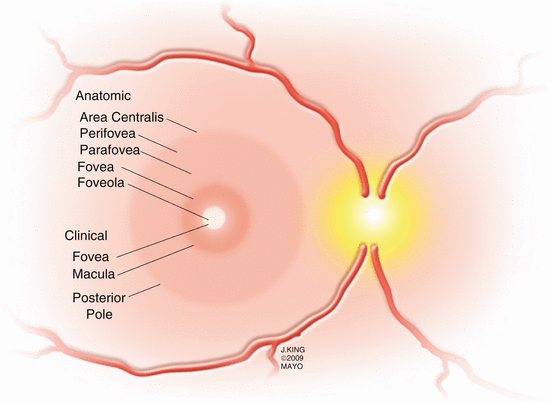
Fig. 2.2
This drawing shows the anatomic and clinical areas of the macula. Despite being referenced more frequently by anatomists, the terms parafovea and perifovea are often used in clinical practice to describe the locations of macular pathology
Beyond the macula, the retina extends past the vortex veins until terminating at the ora serrata. The equatorial diameter of the human eye averages 24.06–24.08 mm, from which the retinal area is calculated to be 1206 mm2 [169, 218]. The ora serrata lies between 5.73 mm (nasally) and 6.52 mm (temporally) posterior to Schwalbe’s line and is characterized by protruding extensions of the retina (teeth) that are separated by indentations of the ciliary epithelium (ora bays) from the pars plana. The strong adhesion between the vitreous and the peripheral retina and pars plana, the so-called vitreous base, extends from 1.8 to 3 mm posterior to the ora bays to 1–2 mm anterior to the ora serrata [150].
A cross-sectional view through the retina just outside the retina centralis shows nine layers (from internal to external): nerve fiber layer, ganglion cell layer, inner plexiform layer, inner nuclear layer, outer plexiform layer, outer nuclear layer, external limiting membrane (ELM), rod and cone inner and outer segments, and retinal pigment epithelium (RPE) (Figs. 2.3 and 2.4). The retina is thickest around the optic disc because of the densely packed nerve fiber and ganglion cell layers, but thickness tapers to 0.18 mm at the equator and 0.11 mm at the ora serrata, since the density of all neural elements decreases in the peripheral retina.
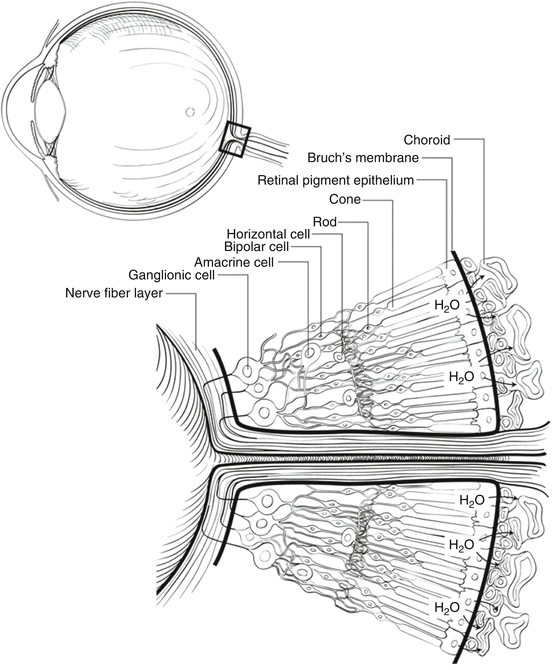
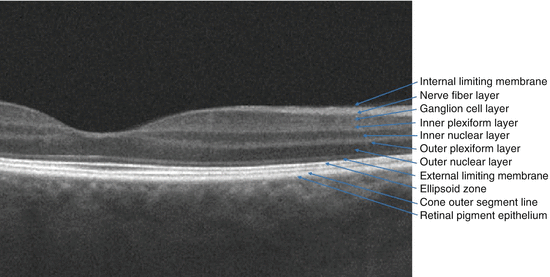
Fig. 2.3
This cross-sectional drawing of the retina near the optic nerve shows the layers of the retina and the locations of important cell types. The pumping mechanism of the retinal pigment epithelium (RPE), which removes water from the subretinal space to keep the photoreceptors in contact with the RPE, is illustrated
Fig. 2.4
The labeling on this spectral domain optical coherence tomography (OCT) scan through thepapillomacular bundle highlights the tissue layers of the retina and important barrier structures. Structures that are commonly identified on the OCT that are often not referred to by anatomists as retinal layers – internal limiting membrane and external limiting membrane – have been included
The layered pattern of cells and connecting neural fibers within the retina enables photic energy to be converted into neuronal signals. Rhodopsin within the outer segments of rods and cones detects incoming photons and converts their energy into an electrical potential by depolarizing adjacent cellular membranes. Photoreceptor cells synapse with dendrites of bipolar cells (first-order neurons) and the processes of horizontal cells (integrating neuronal cells) in the outer plexiform layer. In the inner plexiform layer, bipolar cells synapse with dendrites of ganglion cells (second-order neurons) and amacrine cells (which provide cross wiring). Ganglion cell axons comprise the nerve fiber layer, which aggregate to form the optic nerves until they synapse with third-order neurons in the lateral geniculate body.
The central 0.4 mm of the macula is capillary-free with its cells obtaining their nutrients from the choriocapillaris [54]. The fovea contains long and slender red and green (but not blue) cones that are aligned perpendicular to the RPE, which maximally sensitizes them to incoming photons. Because the fovea lacks the inner nuclear layer, inner plexiform layer, ganglion cell layer, and nerve fiber layer, light scatter is minimized. The external plexiform layer has an unusual configuration as its axons make a right angle turn to assume a course parallel with the retina surface. After coursing a short distance parallel to the retinal surface (while forming Henle’s layer), the axons turn perpendicular to the RPE to synapse with dendrites of overlying bipolar cells outside the central 0.2 mm diameter of the fovea.
Xanthophyll pigments within bipolar and ganglion cells give the retina its characteristic yellow color and function to decrease chromatic aberration, absorb potentially toxic blue light, and scavenge free radicals. Lutein is scattered throughout the posterior pole, whereas zeaxanthin is concentrated in the foveal region [81].
The parafovea and perifovea are frequently referenced regions in clinical practice. The parafovea is a 0.5 mm ring of retina that surrounds the fovea and is characterized by an accumulation of ganglion and inner nuclear cells with a thickened Henle’s layer. The density of cones in the parafovea is lower than within the fovea and rods are beginning to be found. The perifovea is the outermost ring of the retina centralis, between 1.25 and 2.75 mm from the foveal center. The perifovea begins where the ganglion cell layer is four nuclei thick and ends where it thins to a single layer.
The ora serrata is the anterior boundary of the retina where it forms the junction between the multilayered neurosensory retina and the monolayered, nonpigmented epithelium of the ciliary body. The tissue in this region is thin and avascular and enjoys a close relationship with the vitreous base and zonular fibers. Collagen fibrils insert into the internal limiting membrane (ILM) of the retina at the vitreous base, which normally extends from 2 to 4 mm posterior to the ora. The vitreoretinal adhesion is particularly strong along the posterior margin of the vitreous base, which makes this a common site for the development of retinal tears. A gradual loss of the nerve fiber layer, ganglion cell layer, and plexiform layers occurs near the ora as these layers are replaced with neuroglia and Müller cells. Both the ILM, into which vitreous base inserts, and the external limiting membrane (ELM), which continues anteriorly between the pigmented and nonpigmented layers of pars plana, are thickened in this region.
2.2.3 Microanatomy of the Retina Neurons
Cells within the retina fall into one of the three histologic groups: neuronal, glial, and vascular. The intricate interactions between the three types of neural cells, photoreceptors, interneurons, and ganglion cells, give the retina its primary function – converting light energy into electrical impulses. The densely packed rod and cone photoreceptor cells act as the primary neurons in the visual pathway by detecting individual photons to accurately construct visual images. Any change in the axial arrangement of the photoreceptors alters visual perception by producing micropsia if the cells are abnormally separated or metamorphopsia if the alignment is irregular.
Cone photoreceptor cells are comprised of four distinct anatomic portions: inner segments, outer segments that contain the visual pigment, a perikaryal region that contains the cell nucleus, and a synaptic terminal. The three different types of cones each contain only one light-sensitive pigment, which results in three different spectral sensitivities. The blue, green, and yellow cone pigments maximally absorb incident light with wavelengths of 450 nm, 530 nm, and 565 nm, respectively. The visual pigment in rods, rhodopsin, is composed of the light-sensitive chromophore retinol attached to the protein opsin [197] and is most sensitive to light with a wavelength of 500 nm.
The photoreceptor outer segments have two important anatomic connections, to the inner segments (cell bodies) of the photoreceptor cells and to the extracellular matrix that separate the outer segments from the RPE. The acid mucopolysaccharides of the matrix are probably synthesized by the photoreceptor inner segments and form the “glue” that keeps the outer segments attached to the RPE [5]. The nuclei of the cones are situated 3–4 μm internal to the ELM and comprise the outer nuclear layer. The photoreceptors form synaptic junctions with both interneurons and Müller cells. The plasma membrane of each photoreceptor and Müller cell is differentiated into a dense band known as the external limiting membrane. Rods and cones do not come into direct contact as they are insulated from each other by the Müller cells. The Müller cells contact each other in zonula adherens, which are thought to form a relative diffusion barrier between the intercellular space of the inner retina and the extracellular matrix between the photoreceptor outer segments and the RPE.
The outer plexiform layer lies between the inner and outer nuclear layers. The synaptic zone is composed of numerous intercellular junctions and synapses between neural and glial processes and creates the middle limiting membrane. This resembles the ELM and may also act as a partial barrier to diffusion of fluid and larger molecules as it may prevent exudates, hemorrhages, and cysts from spreading throughout the entire retina.
The inner plexiform layer is located between the ganglion and inner nuclear cell layers and contains Müller cell branches, retinal blood vessels, and synaptic processes of the bipolar, ganglion, and amacrine cells. Synaptic units called dyads each consist of a bipolar cell contacting two processes, one from a ganglion cell. Dyad density is approximately 2.9 million/mm2 [47].
The ganglion cell bodies comprise a distinct layer between the inner plexiform and nerve fiber layers. Throughout much of the retina, there is one ganglion cell for every 100 rods and every 4–6 cones, but the ganglion cell to photoreceptor ratio is higher in the macula. This creates a smaller receptor field for each ganglion cell and greater image resolution. There are no ganglion cells at the center of fovea, but the ganglion cells are so densely packed within the extrafoveal macula that there may be two or more for each cone [42]. Ganglion cells consist of two types: midget and diffuse. Midget ganglion cells cover small areas (<10 μm2) and synapse with only one midget bipolar cell, though each midget bipolar cell may synapse with several ganglion cells. The large, polysynaptic, diffuse ganglion cells synapse with retinal bipolar and amacrine cells via dendrites and cells in the lateral geniculate body via axons.
Ganglion cell axons course through the nerve fiber layer of the inner retina until entering the optic nerve. The axons remain unmyelinated until they reach the lamina cribrosa at which point they acquire myelin sheaths. Axons directly contact each other without interposed glial cells, except for interdigitating Muller cell processes. Most axons assume a generally radial course toward the optic nerve except for those immediately temporal to the disc, which form the papillomacular bundle. Since these axons are the first to develop, they form the center of the optic nerve. The nerve fiber layer thickens as axons converge at the optic nerves.
Axons cannot survive if they are detached from their respective cell bodies [152] as both proximal and distal axonal degeneration are seen after acute retinal or optic nerve ischemia. Cotton wool spots or optic disc edema are usually seen on funduscopic examination. Though cotton wool spots were long believed to represent focal infarctions of the retinal nerve fiber layer, they may actually be boundary sentinels of inner retinal ischemia [135]. After axonal degeneration, residual defects in the nerve fiber layer can often be seen on optical coherence tomography (OCT) scanning or funduscopic examination.
Müller cells form tight junctions with both other Müller cells and with neural cells. A continuous row of Müller cell zonula adherens forms the external limiting membrane, which acts as a relative barrier to metabolite movement into and out of the retina [142]. Müller cells comprise the majority of retinal glial cells but astrocytes are more widely distributed between blood vessels and neurons.
2.2.4 Intercellular Spaces
Neural cells are 10–20 μm apart within the retina, with spacing similar to that found in the brain. Intercellular spaces are filled with low density matrix material that permits diffusion of even large proteins [173] until they reach the relatively impervious external limiting membrane. The potential subretinal space is filled with an inter-photoreceptor matrix that is comprised of glycosaminoglycans, glycoproteins, and filamentous structures [85]. The most common inter-receptor matrix protein is interstitial retinal binding protein (IRBP), which is synthesized and secreted by rod photoreceptor cells and binds all-trans retinal and 11-cis retinal. Little is known about other matrix proteins.
2.2.5 Internal Limiting Membrane
The ILM is composed of laminin, basement membrane (BM) proteoglycans, fibronectin, and collagen [96] and represents the retina’s only true basement membrane. The outer portion consists of the basement membrane of the Müller cells, whereas the inner portion is formed by vitreous fibrils and mucopolysaccharides. The ILM is 2000 nm thick over the macula but thins to only 20 nm over the fovea where the density of Müller cells decreases [209]. Müller cell processes form a continuous but uneven border of attachment with the ILM, though the exact nature of the vitreous attachment to the ILM is not known (see Chap. 7).
2.2.6 Circulation
The retina’s demand for oxygen is higher than that of any tissue in the body, and to meet this high metabolic demand, the retina relies on two distinct circulations: the inner two-thirds of the retina relies on the retinal vasculature, whereas the outer one-third relies on the choroidal circulation. Flow through the choroidal circulation is high but variable with easy molecular transfer with the surrounding tissues. Flow through the retinal circulation is low but constant with a high rate of oxygen extraction [11].
2.2.7 Arteries
The central retinal artery (CRA) penetrates the optic nerve about 10 mm posterior to the globe, leaves the nerve at the optic disc, and rapidly branches into progressively smaller arterioles. The central retinal artery supplies the entire inner two-thirds of the retina, except in the 20% of eyes with cilioretinal arteries. The histologic structure of the CRA resembles that of other comparable-sized arteries with a luminal diameter of 200 μm, a wall thickness of 35 μm, a single layer of endothelial cells, a subendothelial elastica, an internal elastic lamina, a medium of smooth muscle, and an external elastic lamina that merges with the adventitia. When the artery enters the eye, the elastic lamina is lost but the muscularis is unusually prominent. Degenerative diseases that affect muscular arteries, such as atherosclerosis and giant cell arteritis, also affect the intraneural retinal artery. Arteries within the retina are spared from giant cell arteritis because they lack an internal elastic lamina, whereas atherosclerosis, with its subendothelial plaque formation and hyperplasia of the intimal and endothelial layers, can affect any portion of the retinal artery.
The retinal artery divides into superior and inferior branches immediately upon entering the eye, then to smaller branches according to either dichotomous (equal-sized bifurcation) or side-arm branching. The arteries are situated in the nerve fiber layer or ganglion cell layers, and only the smaller arterioles descend into the inner plexiform layer to supply capillaries [88]. Strong connections exist between the arteries and cortical collagen in the ILM. Vitreous traction on the ILM can elevate the retinal arteries without affecting deeper retinal layers. The arteries are usually superficial to the veins but may dive to the inner nuclear layer at arteriovenous crossing sites.
Within smaller branches of the artery, the muscular layer thins from seven cell layers at the disc to two layers at the equator, and the luminal diameter thins from 120 μm at the disc to 8–15 μm at the equator. The vascular endothelial cells contain tight junctions that prevent the passage of large molecules into or out of the vascular lumens [167], which limits transfer of materials to diffusion and endothelial pinocytosis. Müller and astrocyte cell processes surround the vessels and insulate them from surrounding retinal neural tissue.
Tissue oxygen tensions, metabolic by-products, and intraocular and systemic blood pressures autoregulate the retinal circulation [52]. It is not known if sympathetic or parasympathetic nerves innervate the retinal arteries, but studies suggest that adrenergic binding sites are present on cells and that retinal blood flow can be altered by adrenergic agonists and antagonists [58, 168].
2.2.8 Veins
The central retinal vein consists of a layer of endothelial cells, subendothelial connective tissue, a medium of mostly elastic fibers, a few smooth muscle cells, and a thin connective tissue adventitia. The vein is separated from the surrounding neural tissue by Müller cells and astrocyte processes. The diameter of the vascular lumen decreases from 150 μm at the disc to 20 μm at the equator. In the peripheral retina, smooth muscle cells in the medium are lost and pericytes take their place. Muscle cells enable the venous diameter to change according to the pressure differential across the lumen. In patients with diabetes or carotid artery disease, sluggish flow relaxes the smooth muscles and the veins become sausage-shaped. The central retinal vein is the only outlet for the retinal circulation, but potential anastomoses between the retinal and choroidal circulations at the disc may become manifest in cases of central retinal vein occlusion or compressive lesions of the optic nerve.
2.2.9 Capillaries
Capillaries are found throughout the retina except in the foveal avascular zone, the retina adjacent to major arteries and veins, and the far periphery. The capillary network originates from the arterioles in the ganglion cell layer and spreads through the inner nuclear cell layer. No vessels are found in the outer plexiform and outer nuclear layers. Capillaries are distributed in two layers with a superficial network in the ganglion and nerve fiber cell layers and a deeper network in the inner nuclear layer. The diameters of these vessels range from 5 to 10 μm. The volume of the outer vascular network is relatively constant throughout the retina, whereas the volume of the inner network varies with the thickness of the nerve fiber layer. Though only one capillary layer is found in the perifoveal region, up to four different capillary layers are found in the peripapillary region.
Peripapillary capillaries drain directly into venules on the optic nerve [84]. Within 2 disc diameters of the nerve, these capillaries have long, straight, or slightly curved paths with minimal anastomoses. This unique anatomy makes the capillaries susceptible to elevated IOP and changes in retinal perfusion pressure, which has been used to explain the development of arcuate scotomas in glaucoma, peripapillary flame-shaped hemorrhages in papilledema and hypertension, and cotton wool spots in disorders causing retinal ischemia.
The capillary wall consists of endothelial cells, pericytes, and a basement membrane. The narrow vascular lumen (3.5–6 μm) and thin endothelial cell bodies cause nuclei to bulge inward, which requires erythrocytes to distort and mold when passing.
The capillary endothelial cells are connected by tight junctions that comprise the inner blood-retinal barrier (BRB) [167]. Pinocytotic vesicles transfer metabolites from the circulation to the retina. Diseases such as diabetes that damage the endothelium and disrupt the BRB allow protein and lipid to leak into the retina. The leakage is reversible through endothelial cell mitosis and the formation of new tight junctions [206].
Pericytes are situated within the endothelial cell basement membranes. Contraction of mammalian pericytes has not been demonstrated in vivo, but pericytes contain contractile proteins and contract in vitro when exposed to endothelin [29], thromboxane A2 [46], and angiotensin II [132]. Loss of pericytes, due to ischemic retinopathies such as diabetes mellitus, weakens capillary walls and results in the formation of microaneurysms [35].
2.3 Hemodynamics, Macular Edema, and Starling’s Law
Movement of fluid both into and out of capillaries depends upon hydrostatic and oncotic pressures. Formation and resorption of macular edema can be described by Starling’s law (Fig. 2.5).
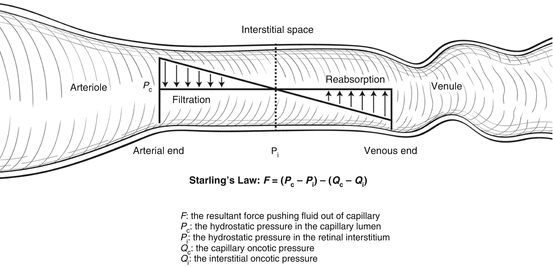
Fig. 2.5
Starling’s law states that movement of fluid across capillary walls is determined by the net hydrostatic pressure less the net oncotic pressure. In eyes with diabetic retinopathy, greater blood flow increases the net oncotic pressure, and breakdown of the blood-retinal barrier enables protein to accumulate in the interstitium and increase the net oncotic pressure. The end result is net fluid movement out of the capillaries into the interstitium leading to macular edema
The four primary Starling’s forces are as follows:
- 1.
Hydrostatic pressure within the capillary lumen (Pc)
- 2.
Hydrostatic pressure within the retinal interstitium (Pi)
- 3.
Capillary oncotic pressure (Qc)
- 4.
Interstitial oncotic pressure (Qi)
Capillary hydrostatic pressure is primarily determined by systemic blood pressure, whereas tissue hydrostatic pressure is approximately the same as intraocular pressure. Most of the capillary oncotic pressure is created by albumin, whereas, with healthy vascular endothelium, tissue oncotic pressure is determined by interstitial proteins. The net force pushing fluid out of capillaries is the difference between hydrostatic pressures and oncotic pressures and can be represented by the following equation:
F = (P c − P i) − (Q c − Q i)
where F is the resultant force that determines fluid movement. If F is positive, fluid moves out of the capillary into the interstitium and forms tissue edema. If F is negative, then the net movement of fluid is out of the tissue and into the capillary. At equilibrium:
F = 0 = ΔP − ΔQ
where there is no net movement of fluid across the capillary walls.
Edema can be defined as the abnormal swelling of soft tissues, which in the case of diabetic retinopathy represents the retinal interstitium. Edema can be cytotoxic, where the fluid accumulates within cells, or vasogenic, where fluid accumulates within the interstitial spaces. Cytotoxic edema occurs with severe ischemic conditions such as central retinal artery occlusions. Starling’s law applies to the formation of vasogenic edema, which is the most common form of edema in retinal vasculopathies such as diabetic macular edema and retinal vein occlusions.
Retinal edema occurs when the net hydrostatic force (forcing fluid into the interstitium) exceeds the net oncotic force (drawing fluid into the capillary lumen) across capillary walls. This usually occurs because of an increase in transluminal hydrostatic pressure, as with systemic hypertension or ocular hypotony, or because of a decrease in transluminal oncotic pressure, as occurs with increased interstitial proteins due to breakdown of the BRB or with a decrease in plasma proteins as with liver disease or protein wasting nephropathies.
Intraluminal pressure falls through the length of the arteriole system, but hydrostatic pressure in the capillaries and venules still depends upon the arterial blood pressure. Systemic arterial hypertension increases capillary hydrostatic pressure and aggravates the severity of diabetic macular edema. Patients with diabetic macular edema should undergo a thorough blood pressure assessment, and if systemic hypertension is discovered, appropriate pressure lowering treatment should be initiated. Reduction of systemic blood pressure may be sufficient to improve diabetic macular edema in some cases [134, 186].
Poiseuille’s law describes flow within a tube, where the resistance to flow is inversely related to the radius of the lumen raised to the fourth power. Retinal arterioles dilate under hypoxic conditions such as diabetic retinopathy, which decreases resistance and increases hydrostatic pressure [178–180]. Retinal blood vessels have been observed to progressively dilate as diabetic macular edema forms [118].
Macular edema also forms in eyes with ocular hypotony. Since tissue hydrostatic pressure is equal to intraocular pressure, hypotonous eyes develop macular edema due to a widened hydrostatic pressure gradient. Edema may improve in these eyes if intraocular pressure rises [115]. When the retina is subjected to vitreous traction, interstitial hydrostatic pressure decreases and macular edema or subretinal fluid accumulates [122].
Capillary oncotic pressure decreases in patients with hypoalbuminemia due to the nephrotic syndrome, protein-deficiency malnutrition, or severe liver disease. On the other hand, breakdown of the BRB allows albumin to leak into the interstitial spaces and increase tissue oncotic pressure. Endothelial damage, like that seen with diabetic retinopathy and vein occlusions, compromises the intercellular tight junctions and breaks down the blood-retinal barrier. Increased vascular porosity results in oncotic pressure shifts and the development of interstitial macular edema. There is a strong correlation between increased vascular endothelial growth factor (VEGF) levels, breakdown of the BRB, and the formation of macular edema [148].
2.4 Biochemical Basis for Diabetic Retinopathy
Patients with DM develop similar microvascular abnormalities in the retinal vasculature, renal glomeruli, and vasa vasorum of the peripheral nerves. Alterations in retinal blood flow and increases in vascular permeability are seen soon after the development of DM. Decreased activity of vasodilators such as nitric oxide together with increased activity of vasoconstrictors such as angiotensin II and endothelin-1 can be measured. The resultant qualitative and quantitative abnormalities in the extracellular matrix contribute to increases in vascular permeability. Microvascular cell loss results from programmed cell death. Cell loss together with the overproduction and deposition of extracellular matrix proteins and periodic acid-Schiff-positive proteins induced by growth factors such as TGF-β leads to progressive capillary occlusion. Chronic hyperglycemia decreases the production of endothelial and neuronal cell trophic factors, which leads to edema, ischemia, and hypoxia-driven neovascularization [25]. In nondiabetic patients, atherosclerosis begins with endothelial dysfunction [131], whereas in diabetic patients initiation seems to involve insulin resistance [90].
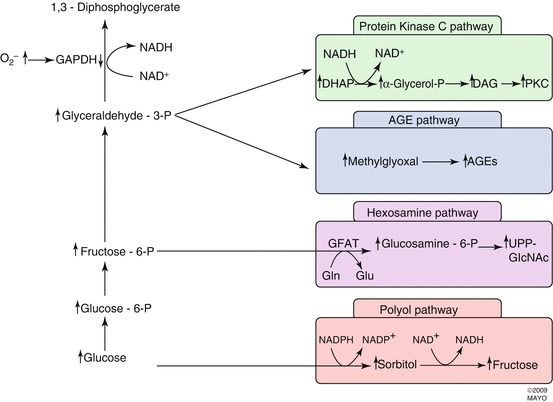
Four hypotheses have been advanced to explain the mechanism of hyperglycemia-induced microvascular damage (Fig. 2.6). These are:
- 1.
Increased polyol pathway flux
- 2.
Accumulation of advanced glycation end products (AGEs)
- 3.
Activation of protein kinase C (PKC)
- 4.
Increased hexosamine pathway flux
Fig. 2.6
Hyperglycemia dysregulates four biochemical pathways: protein kinase C pathway, advanced glycation end product (AGE) pathway, hexosamine pathway, and polyol pathway. Each of the pathways has been associated with the development of diabetic retinopathy
Inhibitors specific to aldose reductase, AGE formation, PKC activation, and the hexosamine pathway each prevent various diabetes-induced abnormalities. No common element to these pathways had been noted until the recent discovery that each causes overproduction of superoxide by the mitochondrial electron transport chain (Fig. 2.7) [25]. Consistent with this observation is that both diabetes and hyperglycemia increase oxidative stress [79].
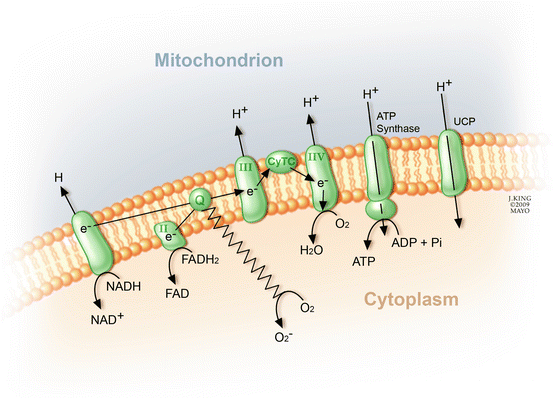
Fig. 2.7
In the presence of O2, electron transfer through the mitochondrial cytochromes regenerates adenosine triphosphate (ATP) from adenosine diphosphate. Dysregulation of each of the previously mentioned biochemical pathways disrupts electron transfer and shunts the electrons toward the synthesis of superoxide (O2 −)
To understand how hyperglycemia increases reactive oxygen species (ROS), one must examine changes in the electron transport within the mitochondria. Hyperglycemia causes overproduction of the electron donors (NADH and FADH2) by the tricarboxylic acid (TCA) cycle [49], which increases the proton gradient across the inner mitochondrial membrane. This sufficiently prolongs the lifespan of electron transport intermediates, such as ubisemiquinone, to generate superoxides. Experimental evidence shows that two regulatory enzymes can be exploited to uncouple hyperglycemia-induced production of ROS. Upregulation of manganese superoxide dismutase (MnSOD) eliminates the production of reactive oxygen species, and an excess of uncoupling protein-1 (UCP-1) eliminates the protein electrochemical gradient [73]. Furthermore, overexpression of either MnSOD or UCP-1 prevents PKC activation of the hexosamine pathway, AGE formation, and an increase in polyol pathway flux. This evidence strongly supports the contention that excessive superoxide is pivotal to the unified theory of diabetic retinopathy.
Other experimental evidence links hyperglycemia, ROS, and the four previously mentioned biochemical pathways. A hyperglycemia-induced increase in ROS decreases glyceraldehyde 3-phosphate dehydrogenase (GAPDH) activity, which increases the concentrations of upstream glycolytic metabolites. These increase flux through the polyol pathway. Elevated triose phosphate levels increase the formation of methylglyoxal-derived AGE, the most common AGE that results from hyperglycemia.
Triose phosphate levels rise due to GAPDH inhibition by ROS [50]. ROS-induced decreases in GAPDH activity increase the concentration of fructose-6-phosphate, the primary substrate for the hexosamine pathway. Inhibition of GAPDH increases concentrations of dihydroxyacetone phosphate, which increases DAG concentrations and activates PKC.
Several experimental models have shown that elevated MnSOD or UCP-1 activity prevents hyperglycemia-induced complications. Overexpression of either protein prevents monocyte adhesion to aortic endothelial cells [25], hyperglycemia-induced decrease in eNOS activity [49], and collagen-induced platelet aggregation and activation [210]. Increased MnSOD activity prevents increase collagen synthesis [37] and decreases programmed cell death induced by hyperglycemia.
Continued oxidative stress leads to chronic inflammation, which is a strong mediator of most chronic diseases including diabetes. Oxidative stress upregulates several transcription factors including AP-1, HIF-1α, β-catenin/Wnt, NF-κB, PPAR-γ, p53, and Nrf2. Activation of these factors leads to the expression of over 500 genes, including those for growth factors, inflammatory cytokines, chemokines, cell regulatory molecules, and anti-inflammatory molecules [156].
Inflammatory cells also produce soluble mediators, such as metabolites of arachidonic acid, cytokines, and chemokines that recruit additional inflammatory cells to the site of damage and produce more reactive species. These key mediators activate signal transduction cascades and induce changes in transcription factors that mediate immediate cellular stress responses. Induction of cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), aberrant expression of inflammatory cytokines (tumor necrosis factor (TNF), interleukin-1 (IL-1), and IL-6) and chemokines (IL-8, CXC chemokine receptor 4 (CXCR4)), and alterations in the expression of specific microRNAs have also been reported to play a role in oxidative stress-induced inflammation [92].
Our current understanding of retinal biochemistry identifies a series of pathways, all of which share substrates and enzymes and are regulated by positive and negative feedback loops. This complicated biochemical mosaic allows for considerable and complicated crossover reactions among pathways, which suggests that successful inhibition of any single pathway will probably produce an incomplete clinical response. If the unified theory of diabetic retinopathy – hyperglycemic production of reactive oxygen species – is correct, then limiting the formation of ROS may ultimately yield the most effective therapies.
A considerable amount of clinical research continues to focus on decreasing diabetic complications by minimizing changes in the four hyperglycemia-driven pathways. Further discussion of these pathways, therefore, appears warranted.
2.4.1 Increased Polyol Pathway Flux
Aldose reductase (the first enzyme in the polyol pathway) has a low affinity for glucose at normal concentrations, but during hyperglycemia glucose is increasingly converted to sorbitol with associated decreases in NADH. Sorbitol is subsequently oxidized to fructose with reconstitution of NADH. Sorbitol oxygenation increases the NADH/NAD+ ratio in the cytosol, which inhibits glyceraldehyde-3-aldehyde dehydrogenase (GAPDH) activity. This increases concentrations of triose phosphate [205], which increases formation of methylglyoxal (a precursor of AGEs) and diacylglycerol (DAG) (which activates PKC). Reduction of glucose to sorbitol consumes NADPH, and since NADPH is also required to reduce glutathione, this could worsen oxidative stress. Unfortunately, attempts to inhibit the polyol pathway in vivo have yielded only mixed results. A 5-year study of polyol pathway inhibition in diabetic dogs prevented diabetic neuropathy but failed to prevent retinopathy [51]. Zenarestat, an aldose reductase inhibitor, had a positive effect on diabetic neuropathy in humans [77].
2.4.2 Advanced Glycation End Products (AGEs)
Intracellular hyperglycemia promotes the formation of AGEs, which are found in elevated concentrations in diabetic retinal blood vessels [182, 184, 185] and glomeruli [89]. The intracellular auto-oxidation of glucose to glyoxal, the decomposition of the Amadori product (glucose-derived 1 amino-1-deoxyfructose lysine adducts) to 3-deoxyglucosone, and the fragmentation of glyceraldehyde-3-phosphate and dihydroxyacetone phosphate to methylglyoxal, each reacts with amino groups of intracellular and extracellular proteins to form AGEs [44, 192, 202]. The AGE inhibitor aminoguanidine partially prevents microvascular damage in animal models [176] and lowers urinary protein concentrations and may slow progression of retinopathy in humans [23].
The intracellular production of AGE precursors damages target cells by modifying critical proteins and altering their functions. These change extracellular matrix components and integrins and modify plasma proteins that bind to AGE receptors. The end result is receptor-mediated production of reactive oxygen species.
The formation of AGEs alters the properties of several extracellular matrix proteins. Advanced glycation end product-induced cross-linking expands the molecular packing of type I collagen and changes the composition of type IV collagen in basement membranes, thereby altering the function of blood vessels [91]. Advanced glycation end product formation on laminin decreases the self-assembly of polymers, decreases binding to type IV collagen, and decreases binding to heparin sulfate proteoglycan [30]. The formation of AGEs on the extracellular matrix interferes with matrix-cell interactions. Modification of type IV collagen-binding domains in the matrix decreases endothelial cell adhesion.
Several cell-associated proteins that bind AGEs have been identified. These include OST-48, 80 K-H, galectin-3, macrophage scavenger receptor type II, and RAGE. These proteins mediate the long-term effects of AGEs on macrophages, glomerular mesangial cells, and vascular endothelial cells through the expression of cytokines and growth factors (interleukin-1, insulin-like growth factor-I, tumor necrosis factor-α, TGF-β, macrophage colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, and platelet-derived growth factor) by macrophages and mesangial cells and the expression of pro-coagulatory and pro-inflammatory molecules (thrombomodulin, tissue factor, and VCAM-1) by endothelial cells. Many of these ligands bind to endothelial AGE receptors and mediate VEGF-induced capillary wall hyperpermeability [129].
2.4.3 Activation of Protein Kinase C (PKC)
The protein kinase C family is made up of at least 11 isoforms, 9 of which are activated by diacylglycerol (DAG). Intracellular hyperglycemia increases DAG synthesis from dihydroxyacetone phosphate in both the retina and renal glomeruli [117]. Hyperglycemia also activates PKC isoforms indirectly by ligating AGE receptors [153] and increasing activity of the polyol pathway [103]. Activation of PKC-β isoforms depresses nitric oxide production and increases endothelin-1 activity to mediate blood flow abnormalities in the retina and kidney [93].
Activated PKC increases the accumulation of matrix protein by upregulating the expression of TGF-β1, fibronectin, and type IV collagen, which is mediated by downregulating nitric oxide [38]. Protein kinase C overexpresses the fibrinolytic inhibitor PAI-1 and activates NF-kB in endothelial and vascular smooth muscle cells [151, 211] and regulates and activates various membrane-associated NADP(H)-dependent oxidases.
A PKC-β-specific inhibitor reduces activity in the retina and renal glomeruli of diabetic animals, reverses diabetes-induced increases in retinal mean circulation time, normalizes glomerular filtration rate, and corrects urinary albumin excretion [116].
2.4.4 Increased Hexosamine Pathway Flux
Excess flux through the hexosamine pathway activates genes that lead to vascular endothelial dysfunction and other changes that are seen in diabetic retinopathy. Excess intracellular glucose in the form of fructose-6-phosphate is diverted from glycolysis in order to provide substrates for reactions requiring UDP-N-acetylglucosamine. End products of these reactions include proteoglycans and O-linked glycoproteins. The mechanism by which increased hexosamine pathway flux mediates hyperglycemia-induced increases in gene transcription is not known, but covalent modification of the transcription factor Sp1 by N-acetylglucosamine (G1cNAc) might explain the link between the hexosamine pathway and hyperglycemia-induced changes in transcription of the gene for PAI-1. The glycosylated form of Sp1 upregulates transcription more than the deglycosylated form. Glycosylated Sp1 is inversely associated with phosphorylated Sp1, which suggests that the two molecules compete for the same binding site and may represent a more generalized mechanism for regulating glucose-responsive gene transcription [82].
2.5 Early Pathophysiology of Diabetic Retinopathy
One of the earliest histopathological changes observed in diabetic patients and diabetic models is thickening of the capillary BM (basement membrane) [66, 67]. Thickening is thought to result from increased synthesis of collagen IV, fibronectin, and laminin and/or reduced degradation of the BM by catabolic enzymes [128, 158, 182, 183]. It remains uncertain whether BM thickening is of primary or secondary importance in the development of DR, but it has been speculated that these matrix modifications may contribute to impaired endothelial-pericyte communication, defects in capillary autoregulation, or inappropriate cell interaction with constituent BM proteins [20, 146, 159, 184].
Mitochondria of retinal pericytes exhibit fragmentation, metabolic dysfunction, and reduced extracellular acidification when exposed to high glucose environments. This may be responsible for the early apoptotic death of both pericytes and smooth muscle cells during DM [67, 138]. The ratio of pericytes to endothelial cells in patients with diabetes decreases from 1:1 to 1:4 [19, 80]. Vascular endothelial cells become dysfunctional at the same time, but over the short term, they can sufficiently compensate for the developing cellular deficit. Because of prolonged exposure to the diabetes-induced biochemical milieu, the replicative ability of the endothelial cells ultimately becomes critically compromised [83, 126].
It has been proposed that the primary cellular defect in diabetic retinopathy lies with the vascular endothelium and that retinopathy could be considered an endotheliopathy [104]. The endothelium is critical to normal function of other cells in the capillary complex [66], and though the precise basis of endothelial, pericyte, and smooth muscle dysfunction in the diabetic retinal microvasculature remains obscure, it is most likely related to an array of cumulative biochemical insults coupled with impaired ability of the cells to repair and renew themselves. Retinal pericytes and smooth muscle cells rely on key growth/survival factors, such as PDGF (platelet-derived growth factor)-B [61, 86], which is selectively depleted during diabetes [36] and is closely related to the formation of acellular capillary casts [80].
2.6 Macular Edema
Retinal nutrition is supplied by two distinct circulations: the choriocapillaris nourishes the outer third of the retina and the two vascular layers (superficial plexus in the axon and ganglion cell layers, deep plexus in the inner nuclear layer) of the retinal capillary circulation. Two distinct, though often coexisting, types of diabetic maculopathy are seen: ischemic maculopathy results from capillary dropout, and macular edema results from breakdown of the blood-retinal barrier in the retinal capillaries and/or RPE.
The exact location of macular edema – extracellular or intracellular – has not been well established as previous studies have implicated both locations. Histologic studies of enucleated eyes with diabetic retinopathy have identified swelling and degeneration of Müller, bipolar, ganglion, and photoreceptor cells [59] and extracellular cystic spaces [69]. Though angiographic ischemia and macular edema may coexist, their effects on visual acuity appear to be independent. Arend [12] found no correlation between blood flow and macular edema, thereby concluding that inner retinal ischemia does not cause macular edema. Retinal thickening appears to correlate with blood-retinal barrier breakdown, but ischemia was not correlated with macular thickening, thereby suggesting that the major cause of retinal thickening was extracellular expansion [174]. These studies conclude that ischemia may lead to a small amount of intracellular swelling, but extracellular fluid accumulation is the primary contributor to macular edema [8].
Even before the development of obvious retinopathy, diabetes can either increase or decrease retinal blood flow. Once retinopathy becomes established, however, retinal blood flow usually increases [171]. The autoregulatory function of retinal blood vessels is lost, and both arterioles and venules are noted to dilate and elongate. This leads to decreases in arteriole resistance and loss of pressure across the length of the arteriole. Effective intraluminal pressure in the retinal capillaries increases, which, according to Starling’s law, increases fluid movement into the extracellular space. The concomitant release of vasoactive cytokines (such as VEGF) from endothelial cells, neuroglia, and activated leukocytes breaks down the blood-retinal barrier via several mechanisms and leads to albumin movement into the interstitium.
Total flow through the choroidal circulation decreases in patients with diabetes [120]. Bulk flow to the choroid is relatively unchanged in diabetics, but indocyanine green angiography shows focal areas of choroidal non-perfusion in eyes with diabetic pigment epitheliopathy. Significant leakage across the pigment epithelium sometimes occurs in eyes that have only minimal retinopathy [200]. Both neovascularization within the choroidal and occlusion of the choriocapillaris occur more commonly in diabetics than in individuals without diabetes. These processes probably result from the same inciting events – basement membrane degeneration and angiogenesis – that lead to the development of diabetic retinopathy [63].
Breakdown of the blood-retinal barrier can be detected by vitreous fluorophotometry before edema accumulates within the retina. This probably results from early protein loss from the gap junctions that allows molecules smaller than albumin to pass out of the capillary lumen. The first manifestation of diabetic retinopathy in other patients, however, may be a retinal pigment epitheliopathy [200]. Retinal pigment epithelial dysfunction is probably underappreciated in patients with DR since overlying retinal vascular disease masks RPE dysfunction on fluorescein angiography.
Histopathologic studies have shown that cystoid macular edema develops in the inner nuclear layer and the outer plexiform layer (Fig. 2.8) [194], but large volumes of fluid can “spill over” to the outer nuclear layer (Fig. 2.9). Both histologic studies and OCT scans suggest the presence of multiple cystoid spaces, but further analysis shows that fluid exists in large cavities that are often spanned by Müller cells.
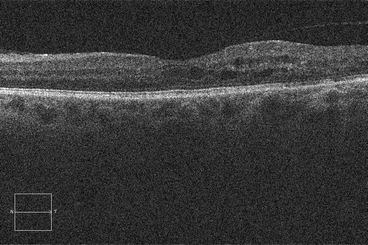
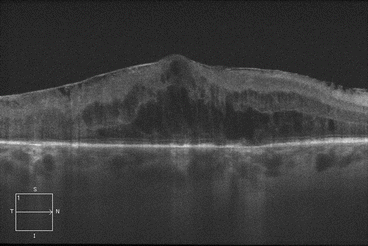
Fig. 2.8
This optical coherence tomography scan demonstrates cystoid macular edema, most which can be seen in the inner and outer nuclear layers
Fig. 2.9
This optical coherence tomography scan through the macula shows significant edema in the outer nuclear layer. Note the prominent epiretinal membrane and the intact external limiting membrane and ellipsoid zone
Fluid accumulation due to leaking capillaries of the inner retina is usually limited by the inner and outer plexiform layers, causing fluid to accumulate within the inner nuclear layer. This fluid displaces tissue in the plexiform layers, increases tissue density, and forms a relative barrier to further spread of the edema. Breakdown of the outer blood-retinal barrier often causes fluid to accumulate between the outer plexiform layer and the external limiting membrane. As demonstrated in horseradish peroxidase studies in diabetic rats [193], eyes with intact external limiting membranes will develop limited exudative retinal detachments with fluid accumulation between the retinal pigment epithelium and photoreceptor outer segments.
2.6.1 Blood-Retinal Barrier
The retina is protected by two distinct blood-retinal barriers: the inner BRB consists of the retinal capillary endothelial cells and their tight junctions, and the outer BRB consists of the retinal pigment epithelium and their tight junctions. The cell membranes of the epithelial and endothelial cells are bilayers with hydrophilic glycerols bounding hydrophobic long-chain fatty acid moieties. Under physiologic conditions, the cell membranes allow small hydrophobic molecules, water, and small uncharged polar molecules to pass, but form an impenetrable barrier to ions and larger uncharged polar molecules.
Adjacent retinal vascular endothelial cells and retinal pigment epithelial cells are connected by tight junctions that are composed of several intercellular proteins including occludin, claudin (23 isoforms), 7H6, cingulin, zonula occludens (ZO)-1,2,3, junction adhesion molecule (JAM), membrane-associated guanylate kinases with inverted domain structures (MAGI)-1,2,3, partition defective genes (PAR)3/6, and multi-pdz protein-1 (MUPP1) (Fig. 2.10) [133]. Some of the proteins have been extensively studied and well characterized, with occludin and the claudins controlling much of the barrier function [106]. Loss of occludin, which is specific to vascular endothelial cells, [10] in diabetic rats correlates with increased blood-retinal permeability to albumin (molecular weight of 66 kD) but not rhodamine-dextran (molecular weight of 10 kD), which may be due to changes in hydrophobicity but not pore size. Claudin-5 prevents passage of small (<0.8 kD) molecules across the blood-brain barrier [141] and probably serves the same purpose in the retina [55]. In retinal capillary endothelial cell tight junctions, ZO-1 is believed to coordinate the intracellular assembly of the junctional complex [53] and is a reliable indicator of overall tight junction function [68]. The junctional adhesion molecules (JAM) facilitate interactions between endothelial cells and leukocytes and assist with tight junction assembly. Junctional proteins may assist with cellular structure since intracellular, circumferential actin bundles aggregate at sites of occludin-positive cell contact, but not at sites that lack occludin.
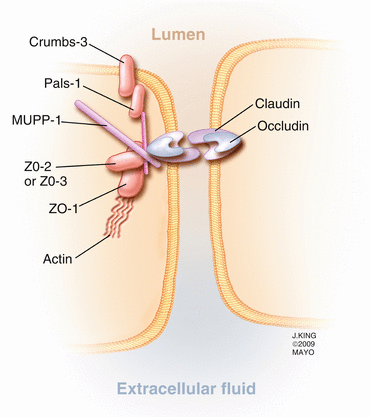
Fig. 2.10
This drawing shows several of the important proteins forming a tight junction between vascular endothelial cells. Note that occludin and the claudins span the intercellular space, while several intracellular proteins, including the multi-pdz protein-1 (MUPP1), support the complex
Transport of ions and molecules across the BRB may be either transcellular or paracellular (Fig. 2.11) [181]. Transcellular passage occurs because of changes in the barrier state or pumping capacity of endothelial cells, whereas paracellular passage occurs because of loss of integrity of the tight junctions. In rat and monkey models of diabetes and in humans, increased fluorescein levels in the vitreous can be detected with vitreous fluorophotometry before fluorescein angiograms show evidence of diabetic retinopathy [41, 97]. These observations suggest that early breakdown of the BRB allows passage of small molecules such as fluorescein but not larger proteins such as albumin, which are necessary for the formation of macular edema.
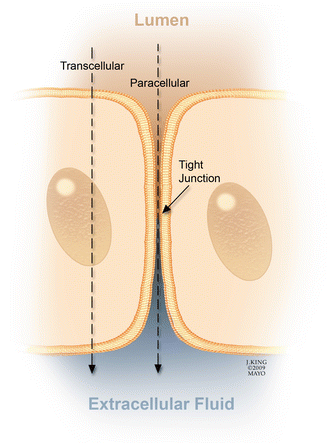
Fig. 2.11
Fluid movement across the blood-retinal barrier (BRB) may be paracellular (through the intercellular tight junctions) or transcellular (across the cell). Vascular endothelial growth factor-mediated breakdown of the BRB promotes fluid movement via both pathways
2.6.2 Biochemical Abnormalities Responsible for Diabetic Retinopathy
During recent years there has been considerable focus on the role of inflammation in the pathophysiology of diabetic retinopathy (Table 2.1) [191]. Increased expression of pro-inflammatory cytokines within the neural retina and upregulation of vascular adhesion molecules that promote leukostasis have been linked to both neurovascular dysfunction and formation of acellular capillaries [98]. Evidence suggests that leukocytes may actively damage the retinal vascular endothelium [123, 189]. Global mRNA expression profiling has detected altered expression of pro-inflammatory cytokines not only in the retinal vessels [72] but also in the neuroglia [26]. Within the diabetic retina, a complex milieu of dysregulated pro-inflammatory factors, including interleukin (IL)-1α, IL-1β, IL-6, and tumor necrosis factor (TNF)-α, develops [191]. Though microglia and infiltrating monocytes are central to inflammatory-mediated central nervous system pathology, the role of these cells in diabetic retinopathy is considerably less well understood. A number of in vitro studies and in vivo investigations of animal models and human postmortem specimens indicate that activation of retinal microglia could play an important regulatory role in diabetes-mediated retinal inflammation [213] by modulating cytokine expression [27] and other pathologic responses [119]. Monocytes and microglia play important roles in retinal homoeostasis, but they are also central to neuro-inflammation, and these have been shown to increase in diabetes, in both humans and animal models [15, 160, 214].
Table 2.1
This table lists several of the chemokines (pro-inflammatory molecules) and cytokines (growth factors) that have been associated with the development of diabetic retinopathy
Chemokines |
Chemokine (C-C) motif ligand 2 (CCL2) |
Endothelin-1 |
Intercellular adhesion molecule-1 (ICAM-1) |
Interleukin 1α (IL-1α) |
Interleukin 1β (IL-1β) |
Interleukin 6 (IL-6) |
Interleukin 8 (IL-8) |
CXCL 10/Interferon induced protein 10 (IP-10) |
Monocyte chemotactic protein 1 (MCP-1) |
P-selectin |
Vascular cell adhesion molecule-1 (VCAM-1) |
Cytokines |
Angiopoietin – 2 (Ang 2) |
Platelet-derived growth factor AA (PDGF-AA) |
Transforming growth factor β (TGF-β) |
Tumor necrosis factor-α (TNF-α) |
Vascular endothelial growth factor (VEGF) |
Very soon after the onset of experimental diabetes in animals, a variety of molecules are upregulated in the retinas and physiological changes are observed. Retinal oxygen tensions decrease before the loss of retinal capillaries in rodent models of diabetic retinopathy [39, 125]. Experimental diabetes in animals results in an increase in retinal VEGF mRNA within the first week, and some authors have concluded that VEGF upregulation is caused by oxidative stress [48, 147]. Upregulation of VEGF correlates with increased expression of intercellular adhesion molecules (ICAM-1), leukostasis, and breakdown of the blood-retinal barrier (BRB) [3]. Pro-inflammatory cytokines such as TNF-α [99] and complement factors [215] are upregulated, and activation of the kallikrein-kinin [121] and renin-angiotensin systems [32] occurs. Each of these changes has been implicated in the pathogenesis of DR, and since some of these pathways are not VEGF dependent, they provide further evidence that inflammation and hypoxia may be independently responsible for DR [4, 43, 45]. Vitreous concentrations of pro-inflammatory cytokines (TNF-α, IL-8, IL-6, VEGF), chemokines (monocyte chemotactic protein-1 (MCP-1)), and other proteins (endothelin-1, sE-selectin, ICAM-1, CXCL10/IP-10) are higher in patients with proliferative diabetic retinopathy or diabetic macular edema than in controls [109].
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


