Introduction
As soon as we are born we begin to die and the end depends on the beginning.
Manilius Astronomica, IV 16
Interest in the connection between development, growth and ageing is not new. Animal studies linking rate of growth and length of life date back to the 1930s. Early findings, however, were conflicting and demonstrated both beneficial and detrimental effects of growth restriction on lifespan. Only later was it understood that timing of the dietary intervention to reduce growth was critical; dietary restriction in later life could slow ageing processes and extend lifespan, whereas a similar intervention instituted in prenatal life was associated with adverse effects.1
Human ageing can be studied at a number of levels, from a population approach down to investigation of individuals at a system or cellular and molecular level. The ageing of a population is defined in terms of mortality statistics, but individual ageing may be defined more broadly as the deteriorative changes with time during post-maturational life that underlie an increasing vulnerability to challenges, thereby decreasing the ability to survive. Progress in understanding why ageing occurs has come from the field of evolutionary biology and there is growing support for the concept that failure of repair is central to ageing. Aetiological studies of ageing have traditionally focused on influences operating in later life, but this has been extended to consideration of the role of early environmental factors, particularly on age-related disease and mortality. More recently, there has been research investigating the developmental origins of system ageing and preliminary studies identifying underlying cellular and molecular changes.
The influence of early nutrition on subsequent health and disease has been a focus of dietary manipulation studies in a range of animal models, but human studies have generally utilized birth characteristics such as size at birth or in infancy as markers of early nutrition and related these to subsequent health outcomes. However, there is also some evidence for the direct effects of early nutrition on long-term human health from natural experiments such as the Dutch Famine Study. This chapter introduces the underlying concept of developmental plasticity and presents evidence for links between birth characteristics and mortality, age-related disease, ageing at a system level (illustrated by the musculoskeletal system) and ageing at a cellular and molecular level. The final section proposes a life course approach to ageing and the relevance of this to clinical practice.
Developmental Plasticity
A series of epidemiological studies demonstrating the relationship between small size at birth and an increased risk of age-related disease in later life led to the developmental origins of health and disease hypothesis.2 Initial observations came from geographical studies. For example, Forsdahl reported that the prevalence of arteriosclerotic heart disease correlated with past infant mortality in the 20 counties of Norway and was the first to suggest that a poor standard of living in childhood and adolescence was a risk factor in heart disease.3 A subsequent study showed that differences in rates of death from coronary heart disease in different parts of England and Wales paralleled previous differences in death rates among newborn babies.4 This led to the Barker hypothesis that adverse environmental influences in utero and during infancy permanently change the body’s structure, physiology and metabolism, increasing susceptibility to disease in later life.
The developmental origins of health and disease are thought to be a subset of the broader biological process of developmental plasticity by which organisms adapt to the environment experienced across the life course. Developmental plasticity involves a single genotype producing alternative forms of structure or function in response to environmental conditions, and it appears to have evolved because it is adaptive. It promotes Darwinian fitness by enhancement of survival and reproductive success through enabling a range of phenotypes better suited to both the prevailing environmental conditions and the anticipated future conditions.5 The biological basis for invoking developmental plasticity as an influence on the risk of disease derives from animal studies in which dietary, endocrine or physical challenges at various times from conception until weaning induce persistent changes in cardiovascular and metabolic function of the offspring. The most commonly used animal models involve a prenatal nutrient imbalance which can be induced by a reduction in overall maternal food intake or by protein restriction in an isocaloric diet. Other models include non-nutritional interventions such as glucocorticoid exposure or placental restriction to impact on early growth and development.
There is growing evidence that epigenetic mechanisms are responsible for tissue-specific gene expression during differentiation and that these mechanisms underlie the processes of developmental plasticity. Examples of epigenetic mechanisms include coordinated changes in the methylation of cytidine–guanosine (CpG) nucleotides in the promoter regions of specific genes, changes in chromatin structure through histone acetylation and methylation and post-transcriptional control by microRNA. Cues for plasticity operate particularly during early development and they may affect a single organ or system, but generally they induce integrated changes in the mature phenotype. In mammals, an adverse intrauterine environment, including suboptimal nutrition, results in an integrated range of responses, suggesting the involvement of a few key regulatory genes that reset the developmental trajectory in expectation of poor postnatal conditions. Mismatch between the anticipated and the actual mature environment exposes the organism to risks of adverse consequences. For humans, it is suggested that prediction is inaccurate for many individuals because of changes in the postnatal environment towards energy-dense nutrition and low energy expenditure, contributing to the epidemic of age-related disease.
Developmental Influences and Mortality
Geographical studies demonstrated that areas with high infant mortality were also the areas with high mortality from coronary heart disease 70 or more years later. However, it took retrospective cohort studies to demonstrate the link between birth characteristics and adult mortality in individuals. This required identification of men and women with historical records of early size. This could then be related to the later occurrence of age-related disease. The first study of this kind was carried out in the county of Hertfordshire, UK. From 1911 onwards, all births were attended by a midwife who measured birth weight. Subsequently, a health visitor went to the baby’s home at intervals throughout infancy and the weight at 1 year was also determined. This information was meticulously recorded in ledgers (Figure 8.1). Many years later, the individuals in these records were traced, enabling the relationship between size at birth or infancy and subsequent health to be investigated.

The associations between birth weight and a range of mortality outcomes have been considered in the Hertfordshire Cohort Study.6 This included 37 615 men and women born in Hertfordshire, UK, between 1911 and 1939, of whom 7916 had died by the end of 1999. For men, lower birth weight was associated with increased risk of mortality from circulatory disease [hazard ratio per standard deviation decrease in birth weight 1.08, 95% confidence interval (CI), 1.04–1.11] and from accidental falls, but with decreased risk of mortality from cancer (hazard ratio per standard deviation decrease in birth weight 0.94, 95% CI, 0.90–0.98). For women, lower birth weight was associated with a significantly (p < 0.05) increased risk of mortality from circulatory disease, injury, musculoskeletal disease, pneumonia and diabetes. Overall, the beneficial effects of an increase in birth weight outweighed the detrimental effects, such that a one standard deviation increase in birth weight reduced all-cause mortality risk by age 75 years by 0.86% for both men and women.
It has been postulated that suboptimal early nutrition underlies the association between low birth weight and increased mortality in adult life. Evidence from the few animal studies that have investigated the effect of dietary restriction during prenatal or early postnatal life, as opposed to post-maturational life, supports this. For example one study of mice dating back to 1920 showed that alteration of diet shortly after birth, sufficient to slow growth, resulted in a shorter lifespan. More recent work using a rat model investigated the effect of a maternal low protein diet on mortality of the offspring. Rat dams were fed either a control diet or a low protein diet from conception until the end of pregnancy. The average lifespan of the female rats exposed to a low protein diet in utero was reduced by 11%7. There was a similar but non-significant trend in the males. Two subsequent studies using rat models also found that that prenatal dietary restriction was associated with a shorter lifespan.
Developmental Influences and Age-Related Disease
The epidemiological studies demonstrating links between weight at birth and adult coronary heart disease mortality have been followed up by cohort studies of survivors where the prevalence of heart disease as well as its risk factors could be identified. Low birth weight has consistently been shown as a risk factor for coronary heart disease and stroke in studies from different populations in Europe, North America and India. The relationship is not limited to the extremes but is graded and linear, operating across the range of normal birth weights. There is some evidence that prematurity is associated with at least some of the risk factors for heart disease but most of the population-attributable risk is thought to be due to reduced fetal growth independent of gestation. Furthermore it appears that the important developmental phases affecting long-term cardiovascular health extend into infancy and childhood.
The association of low birth weight and higher blood pressure is modest, approximately 2 mmHg per kilogram birth weight, whereas the association with frank hypertension is stronger and has been attributed in part to the number of nephrons or the amount of elastin in blood vessel walls, which are lower in individuals born small. There is also evidence that the shape and size of the placenta are important for the subsequent development of hypertension and cardiovascular disease. For example, in the Helsinki Birth Cohort, a small placental surface area was associated with the later development of hypertension in shorter mothers.8 It has been suggested that the effects of placental area on hypertension depend on the mother’s nutritional state. Poor maternal nutrition may compound the adverse effects of small placental size. In better nourished mothers, the placental surface may expand to compensate for fetal undernutrition. They may also reflect effects of the mother’s body size on the transport of nutrients across the placenta.
Small size at birth, in terms of both thinness and low birth weight, has been consistently associated with type 2 diabetes and impaired glucose tolerance. However, in some studies, an inverse J-shaped association has been shown with rates again increasing at the highest birth weights. It has been suggested that gestational diabetes, which is associated with high birth weight, may itself programme long-term adverse health outcomes. Insulin resistance is thought to be a consequence of the term changes in body composition associated with low birth weight, in particular the combination of low lean mass with high fat mass. The relationship between low birth weight and plasma lipids is relatively small: two recent meta-analyses have reported an overall association of approximately 0.04 mmol l−1 higher total cholesterol per kilogram lower birth weight. However, birth weight is only a crude indicator of adverse environmental conditions such as suboptimal nutrition. For example, the Dutch Famine Study showed that famine during early gestation was associated with an atherogenic lipid profile despite being unrelated to birth weight.
Developmental Influences and Ageing of the Musculoskeletal System
Musculoskeletal ageing is a key contributor to the development of physical frailty, disability and dependency in later life. Most aetiological research has focused on identifying adult influences on ageing in the different components of the musculoskeletal system, but recent attention has focused on the contribution of developmental plasticity to ageing in skeletal muscle and bone.
Skeletal Muscle Ageing
Skeletal muscle makes up 40–50% of the human body. It consists of longitudinally arranged muscle fibres broadly classified into two main groups illustrated in Figure 8.2. Type I myofibres have a high capacity for oxidative phosphorylation with high myoglobin levels and mitochondrial density. They are used for endurance activities and are characterized by a slow contraction time and relative resistance to fatigue. Type II myofibres have a greater reliance on glycolytic enzymes. They are used for short, powerful bursts of movement and typically have a fast contraction time but tend to fatigue rapidly. There are multiple changes in muscle with age. Macroscopically there is a reduction in muscle mass with infiltration of adipose and connective tissue. Microscopically there is a reduction in the myofibre number and cross-sectional area, particularly affecting the type II fibres. At the molecular and cellular level, there is decreased muscle protein synthesis with evidence of reduced mitochondrial activity and increased oxidative stress.

The loss of muscle mass and function with age is called sarcopenia and there is increasing recognition of its adverse consequences in terms of increased disability, morbidity, mortality and health care costs. Adult determinants of sarcopenia, including age, gender, size and level of physical activity, have been well described and the beneficial effect of resistance exercise to slow the age-related loss of muscle has been well documented. Twin studies suggest a heritable component and a recent genome-wide association study identified TRHR as an important gene for lean body mass. Nevertheless, there remains considerable unexplained variation in muscle mass and strength between older people which may reflect not only the current rate of loss but also the peak attained earlier in life. Early growth and development of muscle are important determinants of peak muscle mass and strength which are achieved in early adulthood and generally remain stable until the fifth decade. Over the age of 50 years, muscle mass is lost at a rate of 1–2% per year and strength at 1.5–3.0% per year.
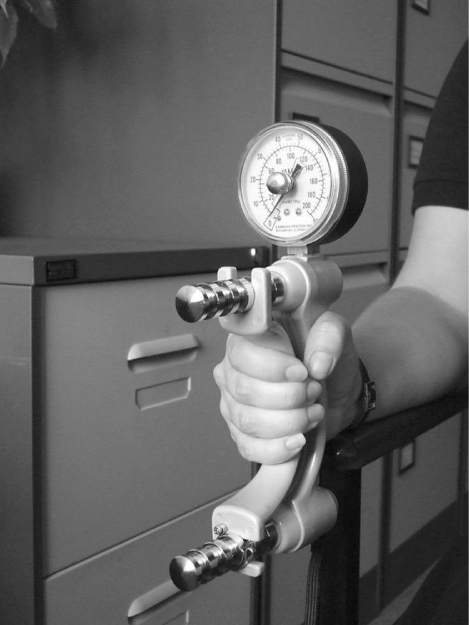
Evidence for developmental influences on ageing muscle has come from a series of observational epidemiological studies linking low birth weight with reduced muscle strength, mass and size in later life. The association between low birth weight and reduced muscle strength was first reported in the Hertfordshire Ageing Study, a retrospective birth cohort study of men and women born in Hertfordshire, UK, between 1920 and 1930 and still living there 60–70 years later.9 They had historical health visitor records of weight at birth and age 1 year and were traced through the National Health Service Central Registry in Southport, UK. Following a home interview, 717 men and women attended a local clinic for measurement of current size and markers of ageing in different body systems, including grip strength measured using hand-held dynamometry (Figure 8.3). Lower birth weight and weight at 1 year were significantly associated with lower grip strength in later life. These relationships remained significant although attenuated after allowing for adult height, suggesting that an adverse early environment may have an effect on both muscle quality and quantity.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


