The Cardiovascular System in Thyrotoxicosis
Irwin L. Klein
Sara Danzi
Almost two centuries ago, Caleb Parry, an English physician from Bath, reported on the association between thyrotoxicosis and the cardiovascular system (1). As the result of over 60 years of clinical and basic science, it is clear that some of the most profound and characteristic symptoms and signs of thyrotoxicosis are those relating to the cardiovascular system (2). Understanding these effects has increased our understanding of the actions of thyroid hormone (3,4). This chapter reviews the molecular, cellular, and organ responses to thyrotoxicosis as it affects the cardiovascular system.
Cardiovascular Hemodynamics
The changes in cardiovascular hemodynamics that occur in patients with thyrotoxicosis are summarized in Table 25.1 (5,6). There are predictable decreases in systemic vascular resistance and diastolic blood pressure, and increases in cardiac output, systolic blood pressure, heart rate, left ventricular ejection fraction, cardiac contractility and mass, and blood volume (6) (Fig. 25.1). While the magnitude of some of the changes varies with disease severity, duration, and patient age, the overall pathophysiology remains the same.
Table 25.1 Cardiovascular Hemodynamics in Thyrotoxicosis | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
 Figure 25.1 Actionsof thyroid hormone on the heart and vascular system. The illustration shows changes in cardiovascular hemodynamics associated with thyroid dysfunction. (From Klein I. Thyroid hormone and the cardiovascular system. Am J Med 1990;88:631, with permission.) |
One of the earliest cardiovascular responses to thyroid hormone administration is a lowering of systemic vascular resistance (7,8), which may decrease by as much as 50% to 70%. The result is an increase in blood flow to the skin, muscles, kidneys, viscera, and heart (5). The decrease in vascular resistance is specific for systemic circulation. It may be caused by a direct vascular action of thyroid hormone (7,8,9), or it may be due to stimulation of vascular endothelial cells to release vasoactive substances such as nitric oxide (9,10). In patients with thyrotoxicosis, β-adrenergic blockade blunts the thyroid hormone–mediated decrease in systemic vascular resistance and the accompanying increase in cardiac output, and administration of vasoconstrictor drugs increases systemic vascular resistance and simultaneously decreases cardiac output (11).
Cardiac contractility is increased in virtually all patients with thyrotoxicosis (2,12,13). Measures of systolic function, such as the rate of increase in ventricular pressure or velocity of contraction, are uniformly increased (12). Measures of diastolic function, including the rate of diastolic relaxation and compliance, also are increased (13), with the exception of that measured in older patients with left ventricular hypertrophy (14).
Cardiac contraction results from the interdigitation of the two major contractile proteins, actin and myosin, requires myosin-catalyzed hydrolysis of adenosine triphosphate (ATP) and has an absolute requirement for calcium (2,4,15,16). One determinant of systolic contractility is the maximum velocity of muscle fiber shortening, which correlates with the inherent ATPase activity of the myosin molecule. There are two isoforms of cardiac myosin, α-myosin and β-myosin; the ATPase activity of the heavy chains of α-myosin is greater than that of the heavy chains of β-myosin. Each heavy chain is encoded by a different gene; the expression of the gene for the heavy chain of α-myosin is stimulated by triiodothyronine (T3) in rodents through an increase in gene transcription and posttranscriptional actions (15,16,17,18). There are substantial species differences in the expression of cardiac myosin and the response to T3. In humans, cardiac muscle obtained at autopsy contains predominantly β-myosin (3), as did the cardiac muscle of a patient with thyrotoxicosis who died suddenly (19). Although thyroxine (T4) therapy increased the content of messenger RNA (mRNA) for the heavy chain of α-myosin in a patient with severe hypothyroidism (20), it is unlikely that thyroid hormone is a major determinant of myosin isoforms or that substantial alterations in myosin gene expression occur in humans with thyroid disease (3).
Calcium release from and reuptake into the sarcoplasmic reticulum of cardiac muscle regulate the rate of ventricular contraction and relaxation. The gene for the cardiac-specific calcium ATPase (SERCA2) that regulates the sequestration of calcium in the sarcoplasmic reticulum during diastole is regulated by thyroid hormone (3,4,16). In addition, the expression of the protein phospholamban, which is a negative regulator of calcium uptake by the sarcoplasmic reticulum, is inhibited by T3 (16). Taken together, these observations could account for the increase in calcium uptake in the sarcoplasmic reticulum and explain the increased rate of development of systolic tension and enhanced diastolic relaxation in the heart of patients with thyrotoxicosis (13,15,16).
Table 25.2 Prominent Symptoms and Signs of Cardiovascular Dysfunction in Patients with Thyrotoxicosis | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Increases in resting heart rate are characteristic of thyrotoxicosis (Table 25.2); more than 90% of patients have resting tachycardia, and many have heart rates of 120 beats per minute or higher (3,12,21,22,23). In addition to the increase in cardiac contractility, there is also an increase in cardiac preload and a decrease in afterload, which combine to cause marked increases in cardiac output both at rest and with exercise (24) (Fig. 25.1). Careful analysis of heart rate regulation in patients with thyrotoxicosis revealed an increase in sympathetic tone and a decrease in parasympathetic tone, the change from normal being greater in the former (25). In addition, thyroid hormone may directly stimulate the sinus node pacemaker (21).
Serum erythropoietin concentrations are increased in patients with thyrotoxicosis, presumably in response to the need to increase peripheral oxygen delivery. Renal sodium reabsorption is increased as a result of activation of the renin–angiotensin–aldosterone system and increases in renal perfusion and renal Na+-, K+-ATPase activity. These changes may
explain the increases in total blood volume, plasma volume, and erythrocyte mass reported in patients with thyrotoxicosis (3,5).
explain the increases in total blood volume, plasma volume, and erythrocyte mass reported in patients with thyrotoxicosis (3,5).
Hyperthyroidism and Thermogenesis
Heat production or thermogenesis is the consequence of the multiple energy transformations that occur as a result of intermediary metabolism in living cells. Homeothermic species like humans must produce more heat than poikilothermic species to maintain the core temperature in all environments which in turn allows for the efficient conduct of enzymatic processes. Metabolic rate, which is measured as oxygen consumption (QO2), is greater in mammals than in reptiles. The sum of the heat generated from the basal energy cost of defending against entropy has been called basal thermogenesis. In turn, some of this is thyroid hormone dependent. When obligatory thermogenesis is not sufficient to maintain body temperature, adaptive mechanisms come into play such that the body defends its temperature by reducing heat dissipation and by recruiting facultative thermogenesis. Shivering is the most acute response. In small mammals, including the human newborn and infant, the main site of non-shivering facultative thermogenesis is the brown adipose tissue (BAT) (26,27,28).
The clinical translation of changes in thyroid state is the classic heat and cold intolerance of patients with hyperthyroidism and hypothyroidism, respectively (29). The thermogenic effects of thyroid hormone encompass those that directly generate the heat and those that provide the fuel for thermogenesis. Thyroid hormone induces heat production by increasing ATP turnover and perhaps by uncoupling the thermodynamic efficiency of the energy generating mechanism. Basal metabolic rate could decrease 30% or more in severe hypothyroidism and increase by similar amounts in hyperthyroidism (30).
Thermogenesis increases energy needs necessitating the provision of extra energy, therefore, thyroid hormone also stimulates food intake. Increased thermogenesis also demands additional oxygen supply to tissues and stimulates cardiac function increasing cardiac performance and output (21).
In the processes of both fuel and oxygen delivery, thyroid hormone interacts with the sympathoadrenal system. Thus, at many levels there is a synergism between catecholamines and thyroid hormone. Catecholamines initiate their effects via cell surface receptors. The α-adrenergic receptors mediate vasoconstriction, and stimulation of type II thyroxine 5′deiodinase (30). The β-adrenergic receptors, on the other hand, mediate a variety of other processes, including cardiac contractility, vasodilation, and the production of metabolic heat. α- and β-adrenergic receptors are coupled to adenylyl cyclase by means of guanosine phosphate–binding proteins. β-adrenergic receptors stimulate the production of cAMP, which in turn activates protein kinase A, leading to the classical downstream effects.
The sympathoadrenal system is under the control of both the hypothalamus and brainstem. The sympathoadrenal system may be activated through both components with strenuous exertion or, as in hypoglycemia, in which the adrenal medulla is stimulated and the activity of the sympathetic nervous system (SNS) is suppressed.
Catecholamines increase thyroxine (T4) to triiodothyronine (T3) conversion in selected tissues (30,31). Therefore the sympathomimetic features of thyrotoxicosis cannot be explained by increased sympathetic activity or epinephrine secretion. In addition, some effects of thyroid hormone are similar to those of sympathoadrenal activation, particularly in the cardiovascular system.
It has been suggested that hyperthyroidism resembles a hyperadrenergic state; however, in the best study to date, there was no evidence to suggest enhanced cardiac sensitivity to adrenergic stimulation (32). Several components of the cardiac myocyte β-adrenergic system are regulated by thyroid hormone, such as the β1-adrenergic receptor, guanine nucleotide regulatory proteins, and adenylate cyclase cardiac–specific isoforms. The increase in the first two components appears to be offset with a decrease in the expression of the catalytic subunits resulting in an overall normal response of cAMP generation to β-adrenergic agonists (2,3). Treatment of hyperthyroidism with β-adrenergic blockade improves many of the cardiovascular signs associated with hyperthyroidism. Heart rate is slowed, but the enhanced diastolic performance is not altered after treatment, which indicates that T3 acts directly on the heart to increase calcium cycling (4,13,16).
Cardiovascular Pathophysiology
Cardiac Effects
T3 regulates the transcription and translation of multiple cardiac genes, and it (and perhaps also T4) may have extranuclear non-genomic actions on cardiac muscle as well (10,33) (Table 25.3). T3 regulates gene transcription by binding to specific nuclear receptors, and the complexes bind to specific DNA sequences [thyroid hormone response elements (TREs)] located on target genes in cardiac myocytes (34,35) (Fig. 25.2). The two isoforms of the T3 nuclear receptors (α1, and β) that bind T3, and the α2 form, that does not bind T3 have been identified in the left ventricular muscle and in isolated cultured cardiac myocytes (see Chapter 8) (15). T3 has direct effects on the rate of gene transcription in cardiac myocytes. These effects occur within 30 minutes and result in an increase in myocyte RNA content and protein synthesis (17). T3 also increases the rate of translation and stability of mRNA in these cells.
Table 25.3 Effects of Triiodothyronine on Cardiac Gene Expression | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||
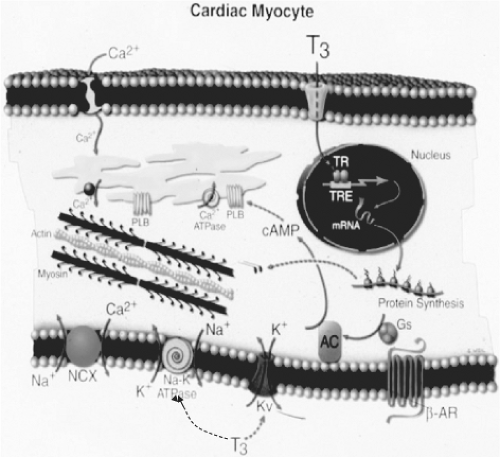 Figure 25.2 Cellular mechanisms of action of triiodothyronine (T3) on cardiac myocytes. T3 binds to T3 nuclear receptors, and the T3 receptor complexes then bind to thyroid hormone response elements of specific myocyte genes, including the genes for α-myosin heavy chains, sarcoplasmic reticulum Ca2+-ATPase, phospholamban, β-adrenergic receptors, adenylyl cyclase, guanine nucleotide–binding proteins, Na+/Ca2+ exchangers, Na+/K+-ATPase, and voltage-gated potassium channels. T3 also has nonnuclear actions on ion channels in the cell membrane. (Reproduced from Klein I, Danzi S. Thyroid disease and the heart. Circulation 2007;116:1725, with permission.) See color plate. |
The different isoforms of the T3 nuclear receptors present in cardiac myocytes may mediate the different genomic actions of T3 in both the atria and ventricles (4,36,37). Indeed, some of the functional and chamber-specific effects of thyroid hormone on the heart may be mediated by a specific receptor isoform. Studies of mice with deletions or mutations of the receptors suggest that the α1 form of the receptor mediates T3 stimulation of heart rate (36), and that the β form is less important as far as the actions of T3 in the heart are concerned (37). It is not clear if this applies to the human cardiac myocyte (2).
Evidence for indirect effects of thyroid hormone on the heart mediated by changes in cardiovascular hemodynamics comes from studies of the cardiac hypertrophic (growth) response to thyrotoxicosis. In humans and animals, chronic thyrotoxicosis causes variable degrees of cardiac hypertrophy (4,38). If
this hypertrophy was caused by a direct effect of thyroid hormone on cardiac protein synthesis, it should not be affected by β-adrenergic blockade. However, both propranolol and bisoprolol block or reverse thyroid hormone–induced cardiac hypertrophy (2,3,39). These results suggest that increased cardiac work is the major mediator of the cardiac growth response in thyrotoxicosis. Further confirmation comes from experiments with heterotopically transplanted hearts in which the heart was exposed to high serum thyroid hormone concentrations without an accompanying increase in cardiac work (38). Under these circumstances, cardiac hypertrophy did not occur, but cardiac gene expression and heart rate increased.
this hypertrophy was caused by a direct effect of thyroid hormone on cardiac protein synthesis, it should not be affected by β-adrenergic blockade. However, both propranolol and bisoprolol block or reverse thyroid hormone–induced cardiac hypertrophy (2,3,39). These results suggest that increased cardiac work is the major mediator of the cardiac growth response in thyrotoxicosis. Further confirmation comes from experiments with heterotopically transplanted hearts in which the heart was exposed to high serum thyroid hormone concentrations without an accompanying increase in cardiac work (38). Under these circumstances, cardiac hypertrophy did not occur, but cardiac gene expression and heart rate increased.
In addition to its genomic actions, T3 has non-genomic or nonnuclear actions on the heart and peripheral vessels (10,33). These actions, because of their rapid time course and lack of associated changes in mRNA or protein synthesis, imply that T3 acts at the plasma membrane, sarcoplasmic reticulum, or mitochondria (33,40). In the plasma membrane of cardiac myocytes, T3, in a concentration of 10 mol/L, increases Na+ ion currents by opening Na+ ion channels. Additional
plasma membrane actions that may be directly regulated by T3 include shortening the duration of action potentials by an effect on voltage-dependent potassium channels and increasing calcium transport in the sarcoplasmic reticulum, nucleotide translocase activity, magnesium flux, and oxidative phosphorylation in mitochondria. These non-genomic actions of T3 may explain, at least in part, the increases in cardiac contractility and cardiac output and decreases in systemic vascular resistance that occur within a few hours in patients given high doses of T3 intravenously after cardiopulmonary bypass surgery (7).
plasma membrane actions that may be directly regulated by T3 include shortening the duration of action potentials by an effect on voltage-dependent potassium channels and increasing calcium transport in the sarcoplasmic reticulum, nucleotide translocase activity, magnesium flux, and oxidative phosphorylation in mitochondria. These non-genomic actions of T3 may explain, at least in part, the increases in cardiac contractility and cardiac output and decreases in systemic vascular resistance that occur within a few hours in patients given high doses of T3 intravenously after cardiopulmonary bypass surgery (7).
Peripheral Circulatory Effects
In patients with thyrotoxicosis of all causes, systemic vascular resistance is reduced from normal values (∼1,700 dynes/sec/cm3) to values as low as 500 to 700 dynes/sec/cm3 (5,6). In animals given T3, the decrease is rapid, occurring before changes in heart rate or cardiac contractility. The decrease in resistance is especially prominent in skeletal muscle, and it also occurs in other organs, including skin, viscera, and especially liver (5,8,11). As systemic vascular resistance declines, so does blood pressure, which in turn causes a reflex increase in heart rate, stroke volume, and cardiac output (Fig. 25.1) similar to that seen with exercise. The reflex nature of these latter changes is supported by the observation that prevention of the decline in systemic vascular resistance with the β-adrenergic antagonist propranolol mitigates or prevents some of the inotropic and chronotropic responses to T3 (11).
As noted above, T3 reduces peripheral vascular resistance through two separate mechanisms (8,10). One is due to a direct action of T3 on arterial smooth muscle tone, as indicated by the ability of T3 to cause relaxation of cultured vascular smooth muscle cells (9). Second, T3 stimulates the synthesis, secretion, or action of vasodilators by endothelial cells (8,10). In rats, synthesis of nitric oxide from L-arginine by nitric oxide synthase in endothelium is stimulated by thyroid hormone (41), and in patients with thyrotoxicosis, increased nitric oxide production contributes to decreased vascular resistance and increased vascular reactivity (8). In addition, the vascular response to acetylcholine, another endothelium-dependent vasodilator, is increased in patients with thyrotoxicosis. In contrast, the vascular response to nitroprusside, an endothelium-independent vasodilator, is not increased, indicating that the endothelium is a specific target for thyroid hormone in mediating these clinically relevant peripheral vascular effects (8).
Thyroid hormone increases the synthesis and secretion of renin and angiotensinogen, the formation of angiotensin II, and the secretion of aldosterone (3,6,42). This activation of the renin–angiotensin–aldosterone system contributes to the increases in renal sodium absorption and blood volume that occur in thyrotoxicosis and the elevation of levels of angiotensin-converting enzyme measured in serum (43).
Thyrotoxicosis can cause hypertension (6,42). This is primarily systolic hypertension, with decreased or normal diastolic pressure and an increase in pulse pressure. The increase in systolic pressure is in part due to decreased arterial compliance, which in turn appears to be the result of tachycardia that is so common in patients with thyrotoxicosis (13,22,24). The prevalence of this finding is estimated at 25% in patients between 30 and 65 years of age (42).
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


