FACTOR VII
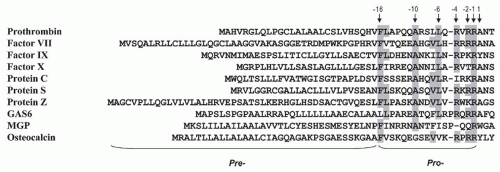
Factor VII is highly homologous in its amino acid sequence and gene organization with the other vitamin K-dependent proteins, including factor IX, factor X, and protein C (
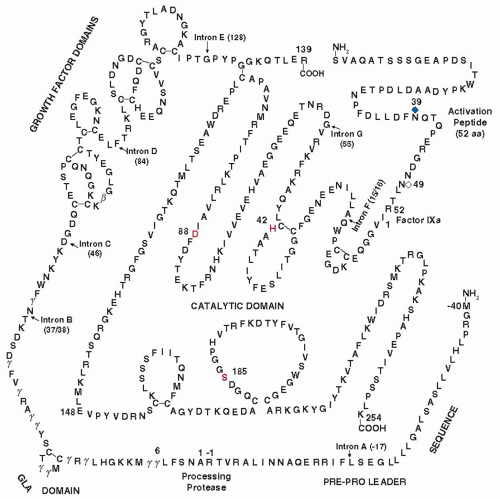
Table 9.1). In the presence of TF, factor VII initiates the extrinsic pathway. Factor VII is a single-chain glycoprotein (Mr 50,000) that is synthesized in the liver and secreted into the blood as a zymogen composed of 406 amino acids (
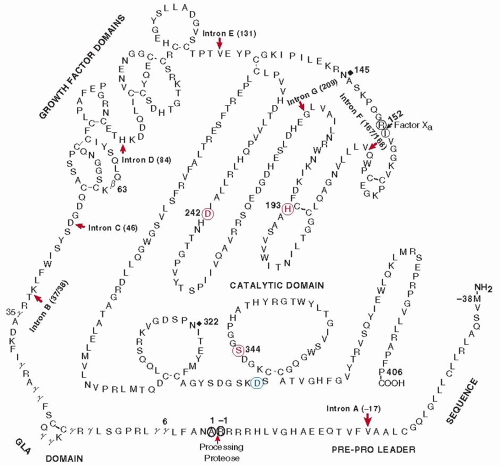
FIGURE 9.2).
2,3,4,5 It contains 10 γ-carboxyglutamic acid residues that are localized in the Gla domain of the protein (amino acids 1 to 35). The Gla region is followed by two epidermal growth factor (EGF)-like domains and a serine protease domain (
FIGURE 9.2). The γ- carboxyglutamic residues require vitamin K for their biosynthesis.
6 Factor VII also contains a residue of β-hydroxyaspartic acid (amino acid 63), located in the first of the two EGF-like domains.
7 The first EGF-like domain stabilizes the factor VIIa-TF complex and participates in calcium binding.
8,9,10 The β-hydroxylation of the Asn63 is not essential for the coagulant activity of factor VIIa since recombinant factor VII is not β-hydroxylated, and functions as well as the plasma-derived counterpart.
11 The first EGF-like domain contains novel
O-linked carbohydrates at residues Ser52 and Ser60.
12,13,14 The Ser52 is
O-glycosylated, being linked to either a disaccharide (Xyl-Glc) or a trisaccharide (Xyl2-Glc) in approximately equal amounts. The Ser60 contains one residue that is conjugated to fucose by
O-glycosylation. Recombinant factor VII/VIIa missing either one or both
O-linked Ser residues exhibits decreased TF binding but has similar calcium binding relative to the wildtype.
9 Factor VIIa has another potential carbohydrate attachment site located at Asn322. Factors VII and VIIa contain two high-affinity calcium-binding sites located in the protease domain and the first EGF1 domain as well as six to seven lowaffinity calcium-binding sites in the Gla domain.
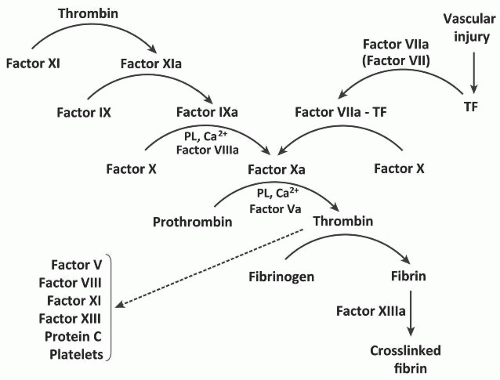
15Factor VII is converted to a serine protease (factor VIIa) by minor proteolysis. Factor VIIa, however, has little if any physiologic activity until it combines with TF. The factor VIIa-TF complex then converts factor X to factor Xa in the presence of phospholipids and calcium ions.
1 Factor VIIa also converts factor IX to factor IXa in the presence of TF and calcium ions.
9 The physiological importance of the latter pathway is unclear.
In vitro experiments evaluating the initiation phase of blood coagulation with ultrasensitive fluorescent markers indicate that factor Xa is generated almost exclusively by the factor VIIa-TF complex during the initiation phase of coagulation.
16 The conversion of human factor VII to factor VIIa is catalyzed by thrombin and factor Xa as well as factor IXa, and factor XIIa.
3,5,17,18,19,20The activation of factor VII is due to the cleavage of a single peptide bond (Arg152-Ile) (
FIGURE 9.2). This leads to the formation of a salt bridge between the α-ammonium group of Ile153 and the carboxylate group of Asp343 and a reregistration of a β-strand in the protein,
21 resulting in a conformational change and formation of an active charge-relay system. Factor VIIa may also be formed by an autocatalytic mechanism,
22,23 but this pathway is very slow and may have
little physiological importance.
24 Factor VIIa is a serine protease composed of a light chain (152 amino acids) and a heavy chain (254 amino acids) held together by a single disulfide bond between Cys135 and Cys262. The light chain contains the Gla domain followed by the two EGF domains, whereas the heavy chain contains the catalytic domain with the active site residues of His193, Asp242, and Ser344. The active site Ser344 is located in the same sequence of Gly-Asp-Ser-Gly-Gly-Pro that is present in all the other serine protease clotting factors, including thrombin, factor IX, factor X, factor XI, and activated protein C (APC).
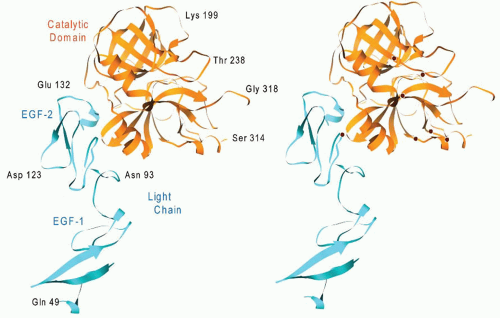
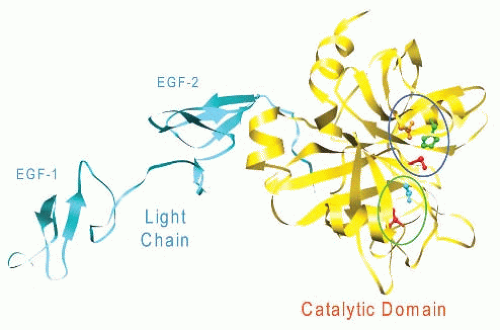
The crystal structure of human factor VIIa lacking the
N-terminal γ-carboxyglutamic acid domain is shown in
FIGURE 9.3.
25 Factor VIIa has an elongated shape similar to that of a tulip, in which the Gla and EGF-like domains form the stem while the catalytic domain forms the blossom.
The cDNA and gene for factor VII have been isolated, and their sequence determined.
26,27 The sequence of the mRNA coding for human factor VII contains approximately 2,450 nucleotides (nts) that code for a prepro leader sequence of 38 amino acids and 406 amino acids present in the mature protein circulating in blood (
FIGURE 9.2). A noncoding region of 1,026 nts plus a poly(A) tail follows the stop codon of TAG. The noncoding region also contains the polyadenylation recognition sequence of AATAAA, which are located 40 nts upstream from the poly(A) tail.
28A second clone has been identified for human factor VII, containing a prepro leader sequence of 60 amino acids.
26 This leader sequence contains an additional 22 amino acids inserted between Val at -17 and Ala at -18 in the 38-amino acid prepro leader sequence encoded by an additional exon in intron A, suggesting that the two mRNA species result from alternative splicing.
27 The removal of the prepro leader sequence requires signal peptidase in addition to a second processing protease that cleaves the peptide bond following the arginine at position -1.
29The gene for factor VII (F7) spans approximately 12.8 kb and consists of eight exons interrupted by seven introns. The positions of the introns with respect to the amino acid sequence and the types of intron-exon boundaries are almost exactly the same as the genes coding for the other vitamin K-dependent proteins (
FIGURE 9.1 and
Table 9.1). The gene coding for factor VII also contains five regions of tandem repetitive sequences, and more than a quarter of the intron sequences consist of minisatellite DNA sequences, which vary in the number of copies among individuals.
30The absence of a CCAAT box in the factor VII promoter contrasts with the promoter structures of factor IX, which has a functional CCAAT box, and factor X, which contains a putative CCAAT box that binds the ubiquitous transcription factor NF-Y.
31,32,33 The major transcription start site for factor VII is located approximately 50-bp upstream from the first initiation Met and is close to the binding sites for a Sp1-like transcription factor and HNF-4.
34,35 The G-C rich Sp1-like site is located at -100 to -94, whereas the HNF-4 site is located at -63 to -58. The factor VII HNF-4 recognition sequence, ACTTTG, is also present in the promoters of factor X and factor IX. A naturally occurring mutation in the factor IX HNF-4 site causes the hemophilia B Leiden phenotype, whereas a similar mutation in the factor VII promoter causes lifelong bleeding and virtually no detectable factor VII.
33,36Genetic defects leading to an abnormal factor VII include a replacement of Arg304 or Arg353 with Gln, resulting in reduced clotting activity,
37,38,39 presumably by a conformational change near the catalytic site. A substitution of Phe38 to Ser (factor VII central)
40 and factor VII variant Gln100 to Arg
41 result in reduced TF binding and impaired activation of factors IX and X.
40 A polymorphism originating from a decanucleotide (CCTATATCCT) inserted at position -323 relative to the first Met-initiating codon
42 and a substitution of G for T at nt -81 disrupts an HNF-4-binding site in the factor VII promoter,
36 resulting in a modest reduction in factor VII levels.
42Pharmaceutical preparations of recombinant factor VIIa have been very helpful in the treatment of patients with factor VIII deficiency and inhibitors as well as in patients with other coagulopathies.
43,44
TISSUE FACTOR
Human TF (Mr 44,000) is a transmembrane glycoprotein synthesized in adventitial fibroblasts. When blood comes into contact with the subendothelium following vascular injury,
45,46,47,48 factor VII binds to TF to form a bimolecular complex in the presence of calcium ions,
1,49,50,51,52,53,54 initiating coagulation. TF is a single-chain protein containing 263 amino acids and is synthesized with a signal peptide of 32 amino acids (
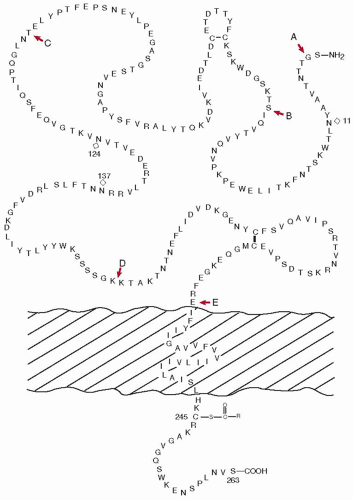
FIGURE 9.4).
55,56,57 The extracellular or cellular surface domain of TF is 219 residues and contains three repeating sequences of Trp-Lys-Ser. It also contains two disulfide bonds linking Cys49 with Cys57, and Cys186 with Cys209.
58 The membrane-spanning region of TF is 23 amino acids (residues 220 to 242), whereas the cytoplasmic portion of the protein at the carboxyl end of the molecule is 21 residues in length. The cytoplasmic portion also contains a half-Cys residue (Cys245) that is acylated by palmitic or stearic acid.
58 Three potential glycosylation sites with a sequence of Asn-X-Thr/Ser (Asn11, Asn124 and Asn137) are also present in the molecule.
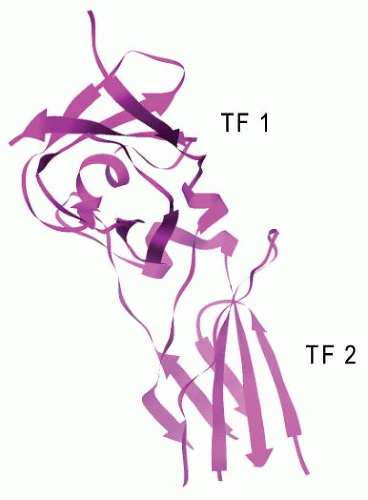
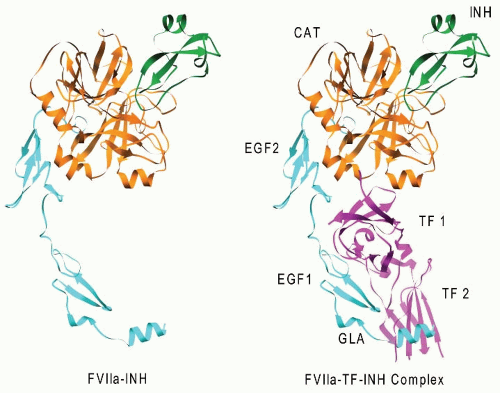
55,56,57The crystal structure of TF,
59 and factor VIIa-TF complex has been determined in several different laboratories.
8,60 The extracellular region of TF (TF-219) consists of two immunoglobulin-like domains that are formed by two antiparallel β-sheets (
FIGURE 9.5). The crystal structure of factor VIIa in a complex with a mutant of bovine pancreatic trypsin inhibitor (BPTI) lacks about two-thirds of the amino-terminal Gla domain of factor VIIa (
FIGURE 9.6,
left). The complex of TF (TF-219) and the factor VIIa-BPTI mutant (
FIGURE 9.6,
right),
60 enhanced by calcium about 150,000 fold,
15 is >100 Å long and 40 to 60 Å wide. In the absence of the inhibitor, the catalytic domain of factor VIIa, bound to the BPTI mutant at the top of the complex, is readily available to bind factor X and convert it to factor Xa.
The mRNA for human TF is 2.3 kb,
55,56,57 including a 5′ noncoding region of 75 bp, 885 bp of coding sequence, a stop codon, and a 3′ noncoding region of 1,141 bp, followed by a poly(A) tail. The gene for human TF (F3) spans 12.4 kb on chromosome 1,
56,61 contains six exons separated by five introns (A to E,
Fig. 9.4), three full Alu sequences, one partial Alu sequence, and a typical TATA promoter element present 26-bp upstream from the cap site. TF in vascular endothelium and monocytes can be induced with several agents, such as phytohemagglutinin and endotoxin, interleukin 1, TNF, phorbol esters, and thrombin.
62,63,64,65,66,67,68 Functional studies indicate that basal TF transcription is controlled by the transcription factor Sp1. A distal enhancer (-227 to -172 bp) containing two AP1 sites and an NFkB site mediates the induction of the TF transcription in monocytic and endothelial cells.
69
FACTOR X
Factor X (Mr 58,800) is a vitamin K-dependent glycoprotein (15% carbohydrate) that is synthesized in the liver and secreted into the plasma. Human factor X is composed of a light chain of 191 amino acids (Mr 16,200) and a heavy chain of 254 amino acids (Mr 42,000) held together by a single disulfide bond (
FIGURE 9.7).
70,71,72,73 The light chain of human factor X contains 11 residues of γ-carboxyglutamic acid, one residue of β-hydroxyaspartic acid (residue 63),
71 and two EGF-like domains,
73,74,75 the first of which contains one high-affinity calcium-binding site.
76 The heavy chain of factor X contains the activation peptide and the catalytic domain.
77 The activation peptide also contains two potential
N-linked carbohydrate-binding sites, including an Asn39-Gln-Thr sequence and an Asn49-Leu-Ser sequence. In bovine factor X, there is an
N-linked (Asn36) as well as an
O-linked (Thr300) carbohydrate chain.
77Factor X is synthesized with a prepro leader sequence that requires two processing steps for its removal (
FIGURE 9.8). These reactions are catalyzed by a signal peptidase as well as by a furin-like enzyme that cleaves the Arg residue on the carboxyl end of the propiece. Additional processing in the single-chain precursor occurs between Arg139 and Ser143, which is the
N-terminal residue in the activation peptide. This protease activity results in the removal of a basic tripeptide of Arg-Lys-Arg and the formation of the two-chain circulating molecule that is held together by a single disulfide bond (
FIGURE 9.7).
The crystal structure of factor Xa lacking the Gla domain is very similar to that of factor VIIa and factor IXa (
FIGURE 9.9), an elongated molecule of 100 Å in length.
78 In the intact molecule,
the Gla region of factor X is partially buried in the phospholipid surface, which is about 61 to 69 Å from the active site.
79Activation of factor X involves the cleavage of an Arg-Ile1 peptide bond in the amino-terminal end of the heavy chain, liberating a small activation peptide of 52 amino acids (
FIGURE 9.7).
70 During the activation reaction, the new amino-terminal Ile1 residue turns and flips into the interior of the catalytic domain. The new α-ammonium group of Ile1 then forms a salt bridge with the carboxylate group of Asp184 (
circled in green,
FIGURE 9.9),
80 generating a serine protease with an active catalytic site involving His42, Asp88, and Ser185 (
circled in blue,
FIGURE 9.9).
The gene for human factor X (F10) contains eight exons and seven introns on chromosome 13 at q32-qter in approximately 27 kb of DNA,
72,81 only 2.8 kb downstream from the gene coding for factor VII.
32 The introns are located between amino acid residues -17 (intron A), 37, and 38 (B), at residue 46 (C), and at residue 84 (D) between the two potential growth factor domains (
FIGURE 9.7 and
Table 9.1). Intron E is located at residue 128 following the second growth factor domain and just before the disulfide bond connecting the light and heavy chains. The last two introns (F and G) are located in the heavy chain or catalytic domain of the molecule and are present between residues 15 and 16 and at residue 55. The rest of the catalytic chain is free of introns.
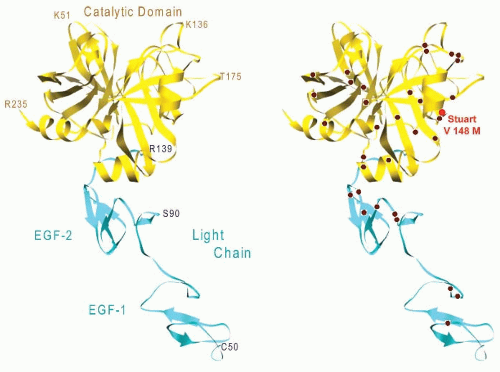
The first patient identified with factor X deficiency
82,83,84 had a single-nucleotide change of G to A (GTG to ATG) resulting in a Val148Met change in the catalytic domain of the protein (
red arrow,
FIGURE 9.8) (Asakai, Roberts, Davie, unpublished data). Other molecular aberrations include partial deletions of exons VII and VIII, and amino acid substitutions of Arg for Gly(-20), Lys for Gla14, Ser for Pro343, and Cys for Arg366.
85,86,87,88,89,90 Amino acid changes that result in a loss of biological activity have been found throughout the catalytic domain as well as the EGF domains (
FIGURE 9.10,
right panel).
The mRNA for human factor X includes 1,475 nts that code for a prepro leader sequence of 40 amino acids, a light chain of 139 amino acids, a connecting tripeptide, and 303 amino acids that constitute the heavy chain.
73,75 The processing or polyadenylation sequence of ATTAAA is unusual in that it is located in the coding sequence and precedes the stop codon by one nucleotide.
28Two transcription start sites, a major one at -16-bp and a minor one at -10-bp upstream from the initiation Met codon, are present in the human factor X 5′-flanking region.
91 Full liver-specific promoter activity is contained within a 457-bp region upstream from the translation start site.
32 An apparent CCAAT sequence, present at -120 to -116 bp, binds the ubiquitous transcription factor NF-Y.
34 An HNF-4 functional element has been localized between -63 and -42 bp, and HNF-4 binding is required for liver-specific expression. Two additional positive regulatory regions have been identified at -215 to -149 bp and at -457 to -351 bp.
32
FACTOR V
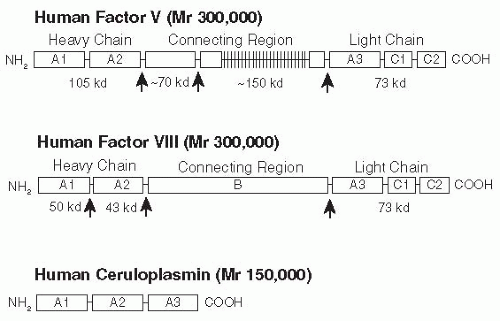
Factor V (Mr 330,000) is a glycoprotein that is synthesized as a single-chain molecule of 2,196 amino acids in liver and megakaryocytes and circulates in blood as an inactive cofactor
at 7 µg/mL.
92 Approximately 20% of the factor V in blood is present in the α-granules of platelets.
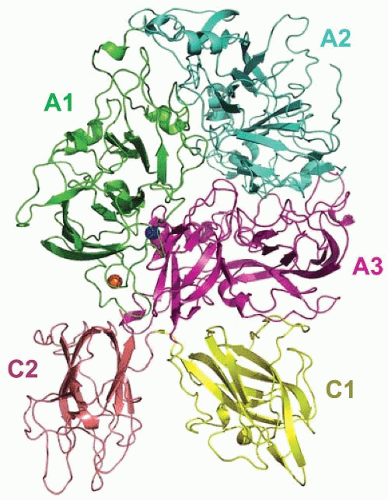
93 Factor V is comprised of six domains (A1-A2-B-A3-C1-C2) that are highly homologous to those in factor VIII and ceruloplasmin (
FIGURE 9.11).
94,95,96,97 The second and third A domains of factor V and factor VIII are separated by a large connecting B domain. In factor V, this domain is 836 amino acids in length, is located between amino acids 710 and 1,545, and has two tandem repeats of 17 amino acids and 31 tandem repeats of nine amino acids with a consensus sequence of [TNP]LSPDLSQT (
FIGURE 9.12). The B domain in factor V shows no similarity in amino acid sequence to that present in factor VIII. Interestingly, chimeric cDNA expression constructs in which the connecting regions of factor V and factor VIII were exchanged show that sequences within the factor V connecting region increase the expression of the factor VIII chimera and its corresponding mRNA by twofold in COS1 cells.
98 In another
study, the connecting region or B domain of factor V did not require the chaperone proteins calnexin or calreticulin for efficient secretion, whereas the connecting region or B domain of factor VIII was required for both chaperone protein binding and proper secretion of factor VIII in a Chinese hamster ovary (CHO) cell line.
99 The third A domain in factor V, as well as factor VIII, is followed by two C domains. Each of the C domains is approximately 150 amino acids in length, with sequence identity of 35% to 50%.
The membrane-binding C2 domain of factor Va contains a β-barrel motif composed of eight antiparallel strands (
FIGURE 9.13),
100 which form two tightly packed β-sheets of five and three strands. The bottom portion of the β-barrel is rich in basic amino acids and consists of three β-hairpin loops that form the calcium-independent membrane- binding site of factor Va.
Before participation in the coagulation cascade, the single-chain factor V undergoes minor proteolysis
95 by thrombin at Arg709, Arg1018, and Arg1545. Factor V can be activated by factor Xa, but the physiological importance of this activation pathway has not been established.
101Factor Va is composed of a heavy chain (A1-A2 domains, Mr 110,000) derived from the amino-terminus of the protein and a light chain (A3-C1-C2 domains, Mr 78,000) derived from the carboxyl-terminus of the molecule (
FIGURE 9.11). The fragments corresponding to the central connecting B region are released from factor V during its conversion to factor Va by thrombin. The newly generated heavy and light chains associate and are held together by calcium that is bound to a high-affinity site
formed when the two chains combine. During the activation of prothrombin by factor Xa, the C1 and C2 domains of the light chain of factor Va are bound to phosphatidylserine in the membranes.
102 Important amino acid residues in the C2 domain of factor Va involved in the phospholipid binding include Trp2063 and Trp2064.
103 Modeling experiments and peptide inhibition studies suggest that the factor Xa binding to factor Va occurs via the A2 domain.
The cDNA for factor V is approximately 7 kb in size and codes for a leader peptide of 28 amino acids and a mature protein of 2,196 amino acids. The 3′ noncoding sequence also contains the typical sequence of AATAAA that functions as a polyadenylation signal.
28The gene coding for human factor V (F5) is located on chromosome 1q21-35 within 300 kb of the genes for the selectin family of leukocyte adhesive molecules.
104,105 The factor V gene spans more than 80 kb of DNA and consists of 25 exons
106 (72 to 2,820 bp) and 24 introns (400 bp to >11 kb) of DNA.
The organization of the gene for human factor V shows remarkable similarity to that of human factor VIII. The factor VIII gene, however, contains one additional exon (i.e., 26 rather
than 25). The gene for factor VIII is also much larger than the gene for factor V, being approximately 180 kb in size. A comparison of the genomic DNA sequences for factor V and factor VIII indicated that 21 of the intron-exon boundaries occur at exactly the same location in the amino acid sequences coded by the two genes.
106 Of particular interest, the connecting B region factors V and VIII are both coded by a single very large exon. In the factor V gene, this exon is located between the 12th and 13th introns, whereas in factor VIII, it is between the 13th and 14th introns.
Recently, a homozygous factor V Leiden mutation (A for G at nt 1,691) has been shown to cause APC resistance and is the major known cause of hereditary thrombophilia.
107,108 This mutation results in the replacement of Gln for Arg at ammo acid residue 506, and this change disrupts the cleavage site for the inactivation of factor Va by APC,
12 and confers a lifelong risk of thrombosis.
109
PROTHROMBIN
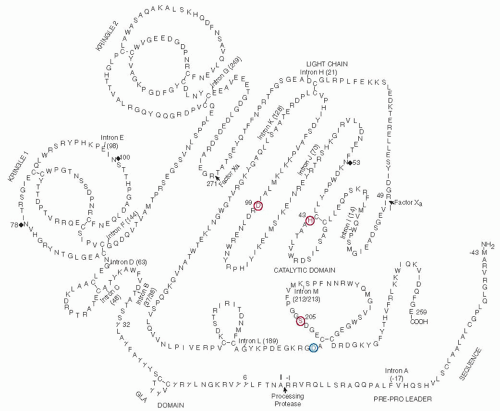
Prothrombin (Mr 71,600) is a glycoprotein containing 8.2% carbohydrate that is synthesized in the liver and secreted into the blood, where it circulates as a precursor to a serine protease at a plasma concentration of 100 µg/mL.
1 The amino acid sequence of human prothrombin is 579 amino acid residues (
FIGURE 9.14).
110,111,112,113,114,115,116 The sequence of human prothrombin
117,118 is identical with about 46% within the heavy chain and has a similarity of about 75% with that of other vertebrates. Prothrombin contains an amino-terminal Gla domain of about 40 amino acids followed by two kringle domains, each containing approximately 80 amino acids and a carboxyl-terminal region with a typical serine protease domain homologous to pancreatic trypsin (
FIGURE 9.14).
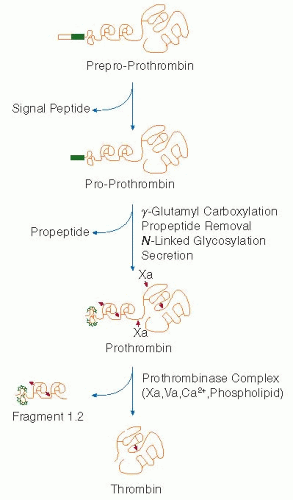
114,119,120During biosynthesis in the rough endoplasmic reticulum, prothrombin undergoes removal of a signal peptide sequence by signal peptidase, carboxylation of amino-terminal glutamic acid residues, cleavage of the propiece by a furin-like proprotein convertase, and the addition of three carbohydrate chains (
FIGURE 9.14). The carboxylation of 10 glutamic acid residues located within the first 40 amino-terminal residues in prothrombin is catalyzed by γ-glutamyl carboxylase, which recognizes the propeptide sequence and is responsible for the vitamin K-dependent conversion of the amino-terminal glutamic acid residues to γ-carboxyglutamate. The γ-carboxyglutamyl residues coordinate Ca
2+ ions, leading to the binding of the Gla region to an anionic phospholipid surface. The addition of the three
N-linked carbohydrate chains occurs at Asn78 and Asn100 in the first kringle domain while the third carbohydrate chain is added to the serine protease domain at Asn53 in the catalytic chain.
111During the final stages of blood coagulation, prothrombin is converted to thrombin. Thrombin is a serine protease composed of a light chain of 49 amino acids (Thr272 to Arg320) and a catalytic heavy chain of 259 residues (Ile1 to Glu259)
(
FIGURE 9.14), held together by a single disulfide bond. Human thrombin undergoes some additional autolysis at the Arg13Thr bond in the light chain, resulting in the removal of a 13-amino acid fragment, reducing the light chain of α-thrombin to 36 residues.
Thrombin generation is due to the cleavage of two internal peptide bonds in prothrombin catalyzed by factor Xa present in the prothrombinase complex.
121 The first cleavage at the Arg320-Ile bond generates a protease called meizothrombin (
FIGURE 9.15), followed by cleavage of the Arg 271-Thr bond forming thrombin and fragment 1.2, a polypeptide containing the Gla domain and two tandem kringle domains. The replacement of the Arg271 by Cys results in a defective molecule (prothrombin Barcelona), demonstrating the importance of this cleavage site.
122In the absence of factor Va, the first cleavage of prothrombin by factor Xa occurs at the Arg271-Thr peptide bond generating fragment 1.2 and prethrombin-2. In a second step, prethrombin-2 is cleaved at Arg320. The catalytic efficiency of the prothrombin-2 pathway, however, is slow and insufficient for physiological clot formation.
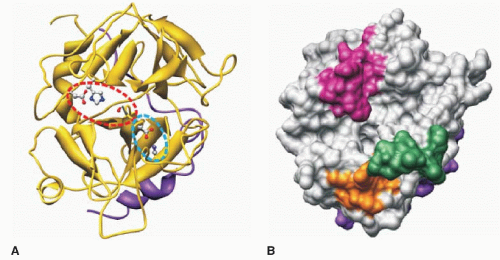
121Thus far, there is no crystal structure available for prothrombin. In 1989, Bode et al.
123,124 published the first detailed crystallographic structures of thrombin and compared its structure with pancreatic trypsin, with which it shares considerable structural similarity. The amino acid sequence of thrombin including the catalytic residues of His43, Asp99, and Ser205 is shown in
FIGURE 9.16. These residues correspond to His57, Asp102, and Ser195 as described in the charge-relay system present in chymotrypsinogen.
125 These three important amino acids (His43, Asp99, and Ser205) are enclosed by a red-dashed oval in thrombin (
FIGURE 9.16A). The His, Asp, and Ser in the catalytic site of thrombin are also located in the middle of an equatorial cleft that separates the adjacent upper and lower β-barrels or hemispheres of a roughly spherical thrombin molecule (
FIGURE 9.16, A,B).
123 The upper lip of the substrate-binding cleft contains the 60 loop
(
magenta shading,
FIGURE 9.16), which is primarily hydrophobic and makes contact with hydrophobic residues present amino-terminal to the scissile bond of the substrate. The lower lip or γ-loop in the substrate-binding cleft (
green shading,
FIGURE 9.16) is more hydrophilic than the 60 loop and plays a role in determining the substrate specificity at the carboxyl side of the scissile bond in the substrate.
Thrombin cleaves peptide bonds immediately following Arg, a specificity that is due to a conserved Asp201 present six amino acids prior to Ser205 and located in the bottom of the S1 primary specificity pocket. This specificity of thrombin is particularly evident for macromolecular substrates and inhibitors and is largely due to two surface regions that are distinct from the catalytic triad of Ser, His, and Asp.
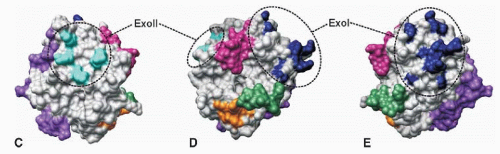
126 These two surface regions are located on opposite ends of the substrate-binding groove in thrombin and have been called the anion-binding exosite I and exosite II (
FIGURE 9.16C-E). Exosite I contains mainly basic amino acids including Lys21, 106, and 107 and Arg62, 68, 70, and 73 (
FIGURE 9.16C-E).
Exosite II (heparin-binding site) consists mainly of the basic amino acids Arg89, 98, and 245 and Lys248 and 252. The sulfated glycosoaminoglycans present in heparin bind to thrombin by an electrostatic interaction and accelerates the rate of thrombin inhibition by antithrombin III.
The cleavage of the Arg320-Ile peptide bond in prothrombin catalyzed by factor Xa results in a new free amino-terminal Ile1 in the catalytic chain of the activated enzyme. The newly generated Ile1 turns and flips into the activation pocket of thrombin where its α-ammonium group forms a salt bridge with the carboxylate group of Asp204 (
dashed blue oval,
FIGURE 9.16A). This Asp is located just prior to the catalytic Ser205. The salt bridge induces a conformational rearrangement in the protein, leading to an active charge-relay system in the serine protease (
dashed red oval,
FIGURE 9.16A), essentially identical to that occurring in
the activation of pancreatic trypsin and all the other coagulation factors that form serine proteases.
The gene for human prothrombin (F2) is present in approximately 21 kb of DNA,
111,112,127,128 and it contains 13 introns (A through M) (
FIGURE 9.14) (84 to 9,447 bp) and 14 exons (25 to 315 bp) located in the coding and 3′ noncoding portions of the gene (
Table 9.2). The first intron (A) is in the prepro leader sequence at residue -17 while the second intron (B) follows the Gla domain between residues 37 and 38 in the mature protein. The third intron (C) is nine residues later at residue 46. These three introns are located in positions analogous to the first three introns in the coding regions of the genes for factor VII, factor IX, factor X, and protein C (
Table 9.1).
27,72,129,130,131 The fourth intron (D) is just before the first kringle, whereas the next intron (E) is present within the first kringle. The fifth intron (F) is located immediately following kringle 1 (residue 144) and the sixth (G) immediately after kringle 2 (residue 249). The seventh intron (H) occurs in the region of prothrombin that becomes the light chain and the remaining five introns (I through M) are located in the catalytic domain at positions 14, 70, 128,
189, and between residues 212 and 213. The sequences at the splice junctions agree with the GT-AG rule of Breathnach and Chambon
132,133 and the consensus sequence of Mount,
133 except for one splice site at the 5′ end of intervening sequence L.
112The gene for prothrombin also contains 30 copies of Alu repetitive sequences, which make up 39% of the gene.
112 This family of DNA sequences is composed of approximately 300 nts, and the human haploid genome contains roughly 350,000 copies of Alu repetitive sequences.
134 In the prothrombin gene, many Alu sequences are tightly clustered and include five sets of tandem repeats. Intervening sequence L (9.5 kb) contains 20 Alu repeats, five of which occur in head-to-tail orientation with no additional DNA between them. The prothrombin gene also contains two copies of partial KpnI repeats (170 bp and 326 bp) located in intervening sequence L.
The mRNA for human prothrombin includes 1,866 nts that code for a prepro leader sequence of 43 amino acids and a mature polypeptide chain of 579 amino acids (
FIGURE 9.14).
111,112 The prepro leader sequence includes a hydrophobic stretch of amino acids (residues -37 to -26) and ends with an arginine residue just before the amino-terminal alanine that is present in the circulating protein. The Arg-Ala peptide bond, however, is not cleaved by signal peptidase.
135 This enzyme cleaves the prepro polypeptide chain near the middle of the prepro leader sequence at one of the small amino acid residues such as alanine cysteine, or serine, leaving a propiece (18 to 24 amino acids) still attached to the polypeptide; the propiece is then cleaved by a second processing protease with a substrate preference for a basic residue at -4, -2, and -1.
29A prepro leader sequence, typical of the vitamin K- dependent coagulation factors (
FIGURE 9.8), plays a role in the carboxylation reaction that occurs on the lumen side of the rough endoplasmic reticulum.
136,137 The conserved Phe and Ala residues at positions -16 and -10 appear to play an important role in the recognition sequence for this carboxylase.
138,139Like the other vitamin K-dependent coagulation factors, the liver-specific expression of prothrombin is transcriptionally regulated. The immediate 5′-flanking sequence does not contain TATA or CCAAT boxes, and the two major transcription start sites are located at -36- and -23-bp upstream of the initiator codon.
140 Full tissue-specific promoter activity is located within 1,000 bp of the transcription start sites.
140,141,142 A weak positive element lies in the region 400-bp upstream of the mRNA coding sequence, accounting for approximately 5% of the total promoter activity in HepG2 cells. A liver-specific enhancer element is located in the region between -940 and -860 bp. DNA protein-binding studies and functional reporter gene analysis with mutant promoter constructs have demonstrated that this 80-bp enhancer contains an HNF-1-binding site flanked by a G-C-rich motif that binds a ubiquitous Sp1-like transacting factor.
140,141,143 All together, six different transcription factors bind to the prothrombin enhancer region and at least three (HNF-4-alpha, HNF-3-beta, and Sp1/Sp3) are important in the regulation of prothrombin expression.
143A common G to A substitution at nucleotide position 20,210 in the 3′-untranslated end of the prothrombin gene results in an elevated level of circulating prothrombin and a 2.8-fo1d increased risk for venous thrombosis.
144,145
FACTOR XI
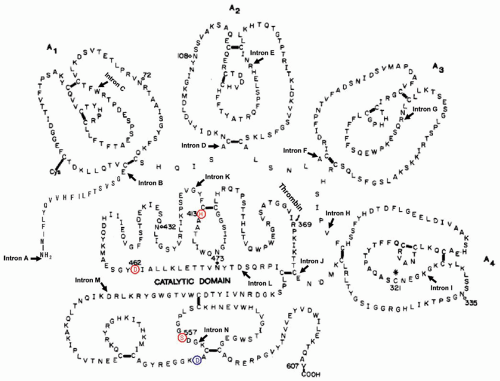
Factor XI (Mr 143,000) is a plasma glycoprotein (5% carbohydrate) composed of 1,214 amino acids synthesized in the liver and secreted into the plasma as a zymogen that circulates as a complex with high molecular weight kininogen (HMWK).
146 Factor XI is an unusual zymogen to a serine protease in that it contains two identical polypeptide chains, each with a catalytic site,
147 linked by a single disulfide bond in the fourth apple domain (Cys321) (
FIGURE 9.17).
148,149 Each of these chains contains four tandem repeats of 90 (or 91) amino acids (“apple” domains) that range in identity from 23% to 34% (
FIGURE 9.17)
150 and are linked by disulfide bonds between the first and sixth, second and fifth, and third and fourth half-Cys.
148 An extra half-Cys (Cys11) present in apple 1 forms a disulfide bond with another half-Cys residue, whereas Cys321 in each fourth apple domain links the two identical polypeptide chains of the protein together by a disulfide bond. The four apple domains in factor XI are also highly homologous to the four tandem repeats present in plasma prekallikrein but have not been identified in any other protein.
151,152 Present evidence indicates that factor XI is bound to HMWK through apple 1, whereas apple 2 is involved in the interaction of factor XI with factor IX.
153,154,155,156 The two catalytic domains in factor XI contain amino acid sequences typical of the pancreatic trypsin family of serine proteases and are located in the carboxyl end of the protein (
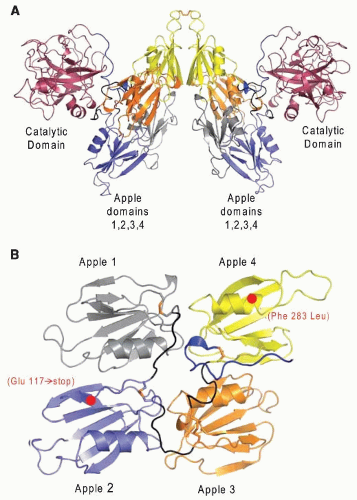
FIGURE 9.17). The four apple domains and the catalytic domains in the dimer of factor XI are readily observed in the crystal structure of the protein (
FIGURE 9.18A,B).
157 Each apple domain (60 × 60 × 20 Å) consists of a single α-helix surrounded by seven antiparallel β-strands. The four tandem apple domains form a shape that resembles a flat saucer. In the dimer, the two saucers are located between the two catalytic domains where they form an inverted V shape.
Factor XIa is a glycoprotein with five potential N-glycosylation sites in each chain. There are four carbohydrate chains, including two on the heavy chain (Asn72 and Asn108) and two on the catalytic or light chain (Asn432 and Asn473), but none on Asn335 of the heavy chain.
The conversion of factor XI to factor XIa catalyzed by thrombin readily occurs in the presence of a negatively charged material such as dextran sulfate, sulfatide, or heparin.
158,159 Factor XI can also be activated
in vitro by factor XIIa in the presence of HMWK and a negatively charged surface.
160 Both of these activation reactions are due to the cleavage of an internal Arg369-Ile peptide bond in each of the two polypeptide chains,
150 resulting in the formation of factor XIa, a serine protease composed of two heavy chains (each 369 amino acids) and two light chains (each 238 amino acids) held together by three disulfide bonds. Each of the two light chains contains a serine protease domain that starts with Ile370. During the activation of factor XI, the newly generated Ile370 turns and flips into the activation pocket where its α-ammonium group forms a salt bridge with the carboxylate group of Asp556. This generates an active serine protease that includes the catalytic triad of His413, Asp462, and Ser557 in the light chain of each factor XIa subunit.
The cDNA for human factor XI has been isolated from a γgt11 expression library prepared from human liver. This mRNA was approximately 2,100 nts long.
150 These data also indicated that factor XI is synthesized as a single polypeptide chain with a typical leader sequence of 18 amino acids (
FIGURE 9.17). Each of the two chains present in the mature molecule contains 607 amino acids. Also, the cDNA for factor XI contains a potential polyadenylation or processing sequence of AACAAA rather than the typical AATAAA.
28 This sequence is located 21 nts upstream from the poly(A) tail and is present in the 166 nts that constitute the 3′ noncoding sequence of the mRNA.
The gene for human factor XI (F11) is approximately 23 kb, located on the distal end of the long arm of chromosome 4 (4q35)
161,162 and contains 15 exons interrupted by 14 introns (
FIGURE 9.17). The first exon codes for the 5′-untranslated region, whereas exon II codes for the signal peptide. The four apple domains are coded by the next eight exons. Each apple domain is coded by two exons interrupted by a single intron, and these introns are located in essentially the same position within each of the four apple domains. The carboxyl-terminal region of factor XIa containing the catalytic chain is coded by five exons, four of which are located in the same positions as those in the genes for human tissue plasminogen activator and human urokinase.
Factor XI deficiency is an unusually mild bleeding tendency that occurs in either sex.
163 Factor XI deficiency is found primarily in the Ashkenazi Jewish population,
164 and mutations occurring almost entirely in two regions of the gene coding for apple 2 and apple 4. The first mutation (type II) results in a stop codon at residue 117 in apple 2, where GAA coding for Gln117 is replaced by a stop codon of TAA (
FIGURE 9.18B), leading to the synthesis of a truncated polypeptide and loss of biological activity. The second principal mutation (type III) results in an amino acid substitution at Phe283 in the fourth apple domain by Leu,
165 resulting in reduced dimerization of the molecule and a lowered secretion.
166 The third, far less common mutation (type I), disrupts normal mRNA splicing and changes a nucleotide sequence of GTAAC to ATAAC at the last intron-exon boundary.
165In a study of 43 patients with severe factor XI deficiency, 49% were due to the type II mutation and 47% to type III.
167,168 More than 152 different mutations have been published that occur in all four apple domains as well as in the catalytic domain.
169