Molecular Biology of the Most Common Primary Central Nervous System Tumors
Brain tumors are thought to arise as the result of genetic and epigenetic alterations that activate oncogenes and inactivate tumor suppressor genes (Figure 40-1 , and see Table 40-1). The precise cell of origin in which the most common brain tumors arise is unknown, but emerging evidence suggests that the malignant transformation of different cell types can give rise to high-grade glioma. There is convincing evidence in animal models of glioma that tumors can arise from the malignant transformation of neural stem cells, 7 very early lineage-specific precursor cells, 8 and more differentiated cells that dedifferentiate 9 and take on key characteristics of precursor cells such as proliferative potential, migratory capacity, and multipotentiality. Identifying the cell type in which tumors arise may provide insight into critical pathologic pathways used by tumor cells. Mouse models of glioma provide strong evidence for the possibility of multiple different cells being targets for malignant transformation, resulting in high-grade glioma.
Significant recent progress has also been made in understanding the cell biology of high-grade glial tumors. An emerging body of data indicates that there is a cellular subpopulation in human glioma, typically consisting of very rare cells, that is distinguished from other tumor cells by the ability to grow in vitro in suspension as neurospheres and to recapitulate the tumor of origin when inoculated orthotopically into immunosuppressed mice. 10 This capacity for tumor initiation is typically not shared with the overwhelming majority of tumor cells, and in contrast to other strategies for immortalizing human glioma, tumor cells grown as neurospheres tend to retain a larger number of the genetic alterations present in the tumors from which they are derived. Study of these tumor-initiating cells, sometimes referred to as tumor stem cells, indicates that they exhibit increased resistance to both radiation and chemotherapy, making them a compelling target for the development of future therapeutic interventions.
Astrocytic Tumors
Diffusely infiltrating astrocytomas are so named because they display morphologic and some biochemical evidence of astroglial differentiation. These are the most common tumors of adults and children, occur throughout the CNS, and exhibit a wide range of histopathologic appearances and clinical behaviors. These tumors are organized by the World Health Organization (WHO) according to tumor grade. 11 Histologically, malignancy is manifested as hypercellularity, cellular atypia, endothelial proliferation, necrosis, and invasion of normal adjacent tissue. WHO grade I tumors are variants of astrocytoma that are generally benign, and WHO grade IV, also known as glioblastoma multiforme (GBM), is the most aggressively malignant. Prognosis is closely associated with pathologic grade. WHO grades II and III exhibit intermediate grades of malignancy, but the evidence of increased mitotic activity in grade III tumors is likely to be a key contributor to poor prognosis. Although low-grade tumors can exhibit a circumscribed growth pattern, all pathologic grades of glioma can exhibit invasiveness, and this characteristic compromises the possibility of treating these tumors with surgery alone. The known propensity of some astrocytic tumors that present as low-grade tumors to progress over time to higher grade tumors has provided insight into the pathogenesis of these tumors, suggesting that they are closely related and that progression is associated with the acquisition of sequential genetic alterations (see Figure 40-1; Table 40-2 ). High-grade astrocytic tumors, GBMs that arise in this manner, are called secondary GBMs. The finding of selected genetic changes (e.g., TP53 mutation) in lower grade tumors that are also present in higher grade tumors along with additional mutations typically found only in higher grade tumors (e.g., epidermal growth factor receptor [EGFR] amplification) suggests that specific genetic alterations are associated with particular pathologic features characteristic of the corresponding grade. Genetic changes currently thought to be important in this regard are shown in Figure 40-1 and Table 40-2, an adaptation of the pathogenesis model first proposed for colon cancer. 12
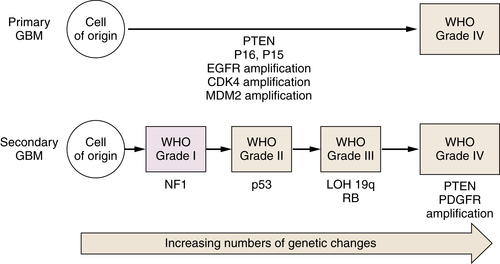
Figure 40-1 Glioblastoma arises in association with the accumulation of multiple genetic alterations. Primary glioblastoma manifests without the presentation of any precursor lesion. Secondary glioblastoma results from the accumulation of additional oncogenic mutations by lower grade astrocytic tumors resulting in increased malignancy. GBM, Glioblastoma multiforme; WHO, World Health Organization.
Mutation of the TP53 gene is likely to be an early event associated with the change of normal cells to low-grade neoplasia. Commonly mutated residues are codons 248 and 273. 13 TP53 has an important role in stabilizing the genome, and the genetic instability resulting from its loss may contribute to the accumulation of multiple mutations in a single cell that are required for the development of highly malignant tumors. Inactivation of TP53 by mutation or epigenetic mechanisms may occur in up to 75% of astrocytomas. MDM2, encoding MDM2, a protein that inhibits the ability of p53 to promote transcription by targeting the protein for degradation, is amplified in approximately 10% of gliomas, and these invariably have a wild-type TP53 gene. A second gene that is also likely to be mutated early in gliomagenesis is IDH1. Mutations of IDH1 were first identified in studies sequencing glioma cell genomes. 14 Mutations of IDH1 are found both in lower grade glioma and in GBM. IDH1 encodes isocitrate dehydrogenase, a Krebs cycle enzyme, and its role in tumorigenesis is an area of intense investigation.
Table 40-2
Tumor Suppressor Genes and Proto-oncogenes Implicated in Brain Tumor Development
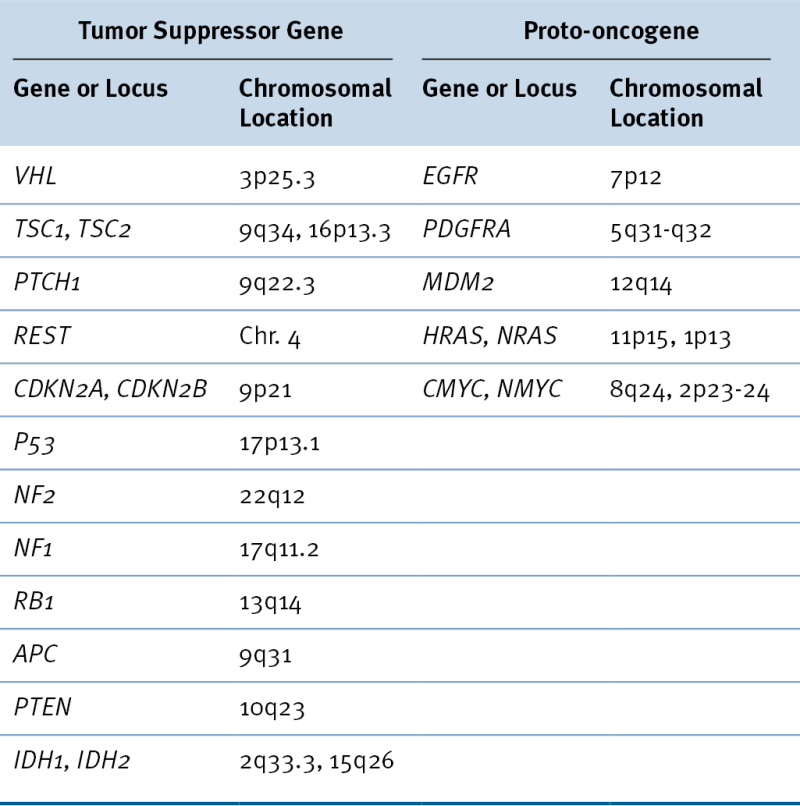
Loss of heterozygosity (LOH) of Ch9p21 at the site of the CDKN2A and CDKN2B loci leads to homozygous deletion of these adjacent genes in approximately 60% of GBM. This deletion results in the loss of the p15 (INK4B), p16 (INK4A), and p14 (ARF) tumor suppressor proteins. CDKN2A and CDKN2B encode cyclin-dependent kinase inhibitors, p16 (INK4A) and p15 (INK4B), respectively, which bind to the cyclin-dependent protein kinases CDK4 and CDK6 inhibiting the catalytic activity of the cyclin D–CDK complex. p1 (ARF) is expressed from a distinct transcript as the result of alternative splicing of the CDKN2A gene and functions to keep MDM2 in the nucleolus so that it cannot degrade p53. Loss of p14 (ARF) results in the enhanced degradation of p53. Other cytogenetic changes including +7p/q, +19q, and –1p/q are widely recognized in high-grade brain tumors, but the best understood is clearly the deletion of chromosome 10, where PTEN is located. The PTEN protein product, PTEN, is a lipid phosphatase that antagonizes the function of the phosphatidylinositol-3-kinase (PI3K) family of lipid messengers and consequently inhibits downstream signaling through AKT1, a serine/threonine kinase that is a key regulator of critical cell functions including cell proliferation and survival.
Several different receptor tyrosine kinases (RTKs) and their cognate ligands have been implicated in the malignant behavior of astrocytic tumors, and especially GBM. These include PDGF/PDGFR, EGF and TGF-α/EGFR, IGF/IGFR, and others. Although amplification of EGFR is found in approximately 50% of GBMs, this change is rarely found in WHO grade III and never in grade I or II tumors. Also, small deletions and rearrangements of the EGFR gene are commonly found in GBM. 15 As many as 50% of tumors in which amplification of EGFR is detectable also have a rearrangement of EGFR in which exons 2 to 7 are deleted. 16 This results in an in-frame deletion that encodes a constitutively active EGFR (EGFRvlll), which, along with EGFR amplification in tumor cells, predicts a poor outcome. Both antibodies and small-molecule therapeutics have been used to target EGFR in glioma, but resistance to such therapies is rapidly manifested when these treatments are used alone. Although members of the PDGF pathway tend to be overexpressed in all grades of astrocytic tumors, amplification and mutation are rare. Activation of RTKs in glioma leads to autophosphorylation of these receptors, which in turn leads to the docking of a series of proteins on phosphorylated sites of the receptors and downstream activation of various effectors. Activation of RAS by the docking protein SOS1 leads to initiation of the RAF/MEK/MAPK (ERK) pathway that leads to the transcriptional activation of genes important for proliferation. Docking of GAB1 to another phosphorylated site on autophosphorylated RTKs leads to activation of PI3K, which in turn initiates a cascade of phosphorylation leading to PDK1 phosphorylation of AKT1. Activated AKT1 in turn phosphorylates both a number of pro-apoptotic proteins, inactivating them, and a series of transcription factors, activating and leading to transcription of other genes important for cellular proliferation. About 80% of GBMs exhibit activation of AKT1, primarily as a result of RTK activation or deletional inactivation of PTEN. 17
Table 40-3
Glioblastoma Multiforme Subtypes as Suggested by Verhaak et al.
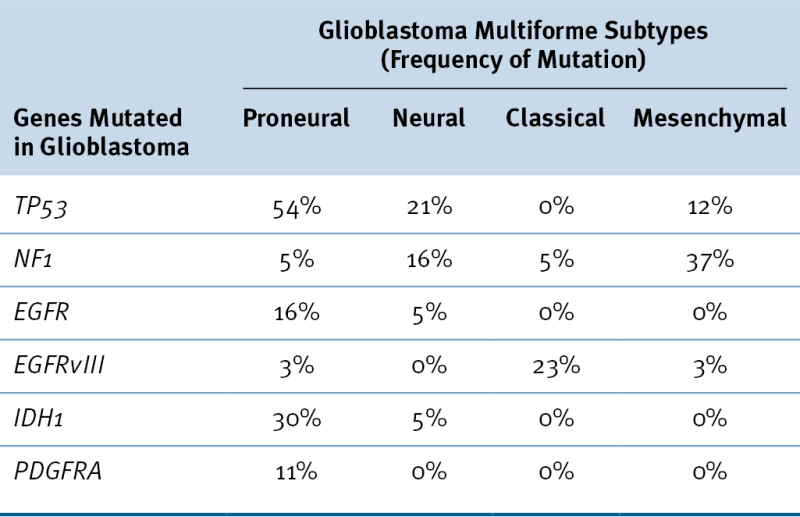
Data from Verhaak RG et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010;17:98-110.
It is now widely recognized that not all GBMs exhibit the same constellation of genetic alterations, and, interestingly, a second clinical presentation of GBM seems to be associated with a distinctive genetic profile (see Figure 40-1; Table 40-3 ). Approximately 90% of patients with GBMs present without evidence of a precursor lesion (de novo, primary GBM). Although high levels of EGFR expression are frequently found in primary GBM, these are rarely detectable in secondary GBM, which is associated with lower grade precursor lesions. In primary GBM, amplification of MDM2 is a more common mechanism for inactivation of TP53 than it is in secondary GBM. The CDKN2A and PTEN loci are also frequently inactivated by mutation in primary GBM, whereas IDH1 is more commonly mutated in secondary GBM.
It has not yet been possible to recognize a characteristic pathology or clinical presentation associated with the genetic heterogeneity that is now well documented to accompany the histologic and cytologic variation characteristic of GBM. Current interpretation of the extensive genomic evaluation of glioma gene structure and gene expression suggests that among histologically indistinguishable tumors, there are definable subtypes of glioma characterized by distinctive molecular alterations (see Table 40-3). 18,19 These classifications may be of prognostic significance, 20 although proven targets for therapeutic intervention have not yet been identified.
Oligodendroglioma
Oligodendrogliomas account for about 20% of primary intracranial tumors and are the second most common glial neoplasm. 1 Most frequently these tumors occur in the cerebral hemispheres, typically within the white matter. They are characterized by various histologic criteria of malignancy, which determine their grade. These tumors can have a highly variable spectrum of differentiation, and their cellular origins are not understood, although it is likely that some high-grade gliomas arise from oligodendrocytic precursor cells (OPCs) that are present in the adult nervous system. 21 There is frequent confusion over the proper diagnostic classification for tumors that do not display prototypical characteristics of cells arising in the oligodendroglial lineage or contain multiple populations of cells, one of which will correspond to the oligodendroglial lineage and the other to the astrocytic lineage. Histopathologically, tumors of the oligodendroglial lineage exhibit round, homogeneous, compact cells with a dense central nucleus and clear cytoplasm, sometimes referred to as a “fried egg” appearance. There is rarely identifiable evidence of mitosis. When tumors are highly anaplastic with obvious necrosis and mitotic figures, they are indistinguishable from GBM and are designated as such. Different histologic grades of oligodendroglioma have been associated with specific patterns of gene expression 22 and genetic alterations. 23
The cytogenetic and molecular biologic profiles of oligodendroglioma are not well characterized but are quite distinct from those of astrocytoma. 23,24 Cytogenetic studies of oligodendroglioma may reveal loss of chromosomes 9p and 22 or gain of chromosome 7, but the most characteristic cytogenetic findings in oligodendroglioma are loss of chromosomes 1p36 and 19q13.3. The presence of these alterations is associated with an enhanced responsiveness to cytotoxic therapies and an improved prognosis. 25 LOH for chromosome 9p, observed in some oligodendrogliomas, corresponds to deletions at the CDKN2A locus, and these have been associated with a poor prognosis for patients with this tumor type. Studies looking for mutations that are frequently found in astrocytic tumors have not detected commonly occurring changes in the TP53 locus. Similarly, Ch10 losses and EGFR amplification have been detected only rarely in these tumors. These distinguishing molecular genetic differences are consistent with more recent genomic scale expression analyses that reveal clear differences between astrocytoma and oligodendroglioma.
Meningioma
Meningiomas account for approximately 20% of primary intracranial malignancies 1 and are derived from mesoderm, probably from cells giving rise to the arachnoid granulations. These tumors are usually benign and attached to the dura mater. They infrequently invade the brain parenchyma. Meningiomas may be found incidentally on a computed tomography (CT) or magnetic resonance imaging (MRI) scan, or they may present with a focal seizure, a slowly progressive neurologic deficit, or symptoms of raised intracranial pressure. Total surgical resection of benign meningioma is curative, and when tumor persists, external beam radiotherapy or stereotaxic radiosurgery reduces the recurrence rate to less than 10%. No effective chemotherapy for the treatment of benign meningioma is known, and there is no curative therapy for anaplastic, highly invasive meningioma that cannot be treated locally.
Meningiomas are an important manifestation of neurofibromatosis type 2 (NF2), which occurs as the result of inactivating mutations of the NF2 gene at 22q12. NF2 functions as part of a molecular complex linking the cellular plasma membrane to the cytoskeleton, where it has been implicated in the regulation of cellular proliferation. About half of NF2 patients develop meningioma, and portions of the NF2 gene are lost in approximately half of sporadic meningioma. The frequency of NF2 mutation does vary somewhat in different histologic types of meningioma. 26,27 NF2 mutations occur in only about 25% of meningothelial meningiomas, whereas more than 75% of fibroblastic and transitional meningiomas have been found to have evidence of NF2 inactivation. Other karyotypic abnormalities are also seen in grade II (atypical meningioma) and grade III (anaplastic meningioma). In anaplastic meningioma, the most frequent cytogenetic abnormalities are deletion of Ch1p, partial or complete loss of Ch10q, and loss of Ch14. Unstable chromosome alterations including rings, dicentrics, and telomeric associations also have been observed.
Medulloblastoma
Among children with cancer, only leukemia is more common than primary brain tumors. Medulloblastoma accounts for approximately 15% to 20% of all pediatric brain tumors and for about 80% of childhood primitive neuroectodermal tumors (PNETs) of the CNS. Only gliomas are more common in children. In contrast to other PNET, these tumors are found in the cerebellum, where they are thought to arise from neural progenitor cells (NPCs) of the fetal external granular layer. 28 The 2007 WHO classification of CNS tumors recognized several histologic subtypes of medulloblastoma, 11 and preliminary studies suggest that these are of prognostic significance. Atypical teratoid/rhabdoid tumors of the CNS identified by somatic mutations of the INI1 gene often occur in the posterior fossa and can be difficult to distinguish from medulloblastoma. In contrast, medulloblastoma is readily distinguishable from childhood glioma in that it appears as a highly cellular, small, round-cell tumor with frequent mitoses and pseudorosettes. Also, medulloblastoma is more responsive to treatment than glioma, as evidenced by the importance of therapeutic response as a prognostic factor in this disease. 29 The 5-year disease-free survival rate approximates 50% to 70%. 30
As noted in Table 40-1, the occurrence of medulloblastoma in three inherited cancer syndromes—Turcot syndrome, Li-Fraumeni syndrome, and Gorlin syndrome—has called attention to the possibility that the genes responsible for these syndromes, APC, TP53, and PTCH1, respectively, or the pathways in which they are active might play a role in sporadic medulloblastoma. Although evidence for APC inactivation in sporadic medulloblastoma has not been identified, TP53 mutations and mutations of PTCH1 and genes in the PTCH1-mediated sonic hedgehog (SHH) pathway are found in sporadically occurring medulloblastoma. 31 Deletions of Ch6q and Ch16q have also been noted in medulloblastoma tumor tissues, and infrequent amplification of MYC and MYCN has been detected. NTRK3 neurotrophin receptor expression in medulloblastoma has been reported to be associated with a favorable outcome, 32 a finding consistent with emerging evidence that medulloblastoma tumor tissue expresses a number of genes associated with the neuronal lineage and that expression profiling of these genes may provide a particularly effective approach to devising clinically useful prognostic biomarkers. 33,34
Molecular Pathophysiology of Primary Brain Tumors
Brain tumors present therapeutic challenges that reflect their occurrence within the closed space of the skull and within a tissue that is particularly sensitive to disruption of its normal function. As a result, simple growth as a space-occupying lesion is particularly problematic in the development of brain tumors as is the invasion of normal tissue, which can occur early in the pathogenesis of some tumor types such as primary GBM and much later in others. The proliferation of brain tumors is thought to be the result of the deregulation of oncogenes and tumor suppressor genes as described previously, and many of the growth-stimulatory pathways that contribute to tumorigenesis in the CNS have been extensively examined. In addition to the oncogenetic alterations described for individual tumors, important regulatory roles in the proliferation of brain tumors include those mediated by EGF and PDGF, which are widely expressed in the normal brain as well.
Glioma and medulloblastoma are typically highly invasive. This characteristic is thought to play an important role in their recurrence after treatment, which tends to occur locally at their original site of presentation. The mechanisms that underlie glial tumor cell invasion are an active area of research. 35–37 Clinicians observed long ago that such malignant tumors have preferred routes of invasion along white matter tracts—although little is known of the manner in which tumor cells recognize white matter tracts and migrate along them. Integrins, especially AvB3 and AvB5, seem to play an important role in this process. 38 Degradation of the extracellular matrix by proteinases produced by tumor cells is now recognized as an important early step in the invasion of normal tissue. Glial tumor cells express urokinase-type plasminogen receptor (UPAR) on their surface, 39 which binds to urokinase plasminogen activators (UPAs) that are also highly expressed in the most malignant gliomas. 39 UPAR presents activated UPA to the extracellular matrix (ECM), where plasminogen is cleaved to active plasmin. This degrades ECM constituents including fibronectin, laminin, and proteoglycans that are especially prominent in the ECM surrounding blood vessels. Matrix metallopeptidases, especially MMP2 and MMP9, also seem to play important roles in ECM degradation. 40 Interestingly, some brain tumors produce inhibitors of various plasminogen activators, thereby inhibiting ECM degradation. The extracellular milieu of the CNS also contains high concentrations of glycosaminoglycan, hyaluronan, and chondroitin sulfate, which are detected in only low amounts in other tissues. How brain tumor cells move through a matrix of such molecules is not known.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


