Systemic vascular thrombosis |
DVT of lower extremity, thoracic, abdominal, or pelvic veins, pulmonary embolism, arterial thrombosis with downstream ischemia and infarction |
Reproductive manifestations |
Recurrent fetal loss, preeclampsia, abruption placentae, miscarriage, prematurity, intrauterine fetal demise, intrauterine growth restriction, and oligohydramnios |
Neurologic manifestations |
Stroke, TIA, migraines, seizures, chorea, Guillain-Barrè Syndrome, transient global amnesia, dementia, diabetic peripheral neuropathy, and orthostatic hypotension |
Cardiovascular manifestations |
Thrombotic occlusion of nonatherosclerotic coronary artery, thrombotic myocardial infarction, coronary artery disease, premature atherosclerosis, atherosclerotic myocardial infarction, peripheral vascular disease, valvular abnormalities |
Pulmonary manifestations |
Pulmonary hypertension attributable to thrombotic occlusion, other pulmonary hypertension, acute diffuse alveolar damage |
Hepatic and gastrointestinal manifestations |
Esophageal necrosis with perforation, intestinal ischemia/infarction, pancreatitis, colonic ulceration, acalculous acute cholecystitis attributable to biliary vascular occlusion, mesenteric venoocclusive disease, primary biliary cirrhosis |
Renal manifestations |
Thrombosis of renal arteries or veins, renal infarction, APS nephropathy, renal artery stenosis, minimal change disease/focal segmental glomerulonephritis, membranous nephropathy, mesangial C3 nephropathy, pauciimmune crescentic glomerulonephritis |
Skin manifestations |
Ulcerations attributable to microvascular infarction, livedo reticularis, skin ulcerations, skin necrosis, necrotizing vasculitis, livedoid vasculitis, thrombophlebitis, erythematous macules, purpura, ecchymoses, painful nodules, subungual splinter hemorrhages, Anetoderma, discoid lupus erythematosus, cutaneous T-cell lymphoma |
Retinal abnormalities |
Retinal vein occlusion, cilioretinal artery occlusion, optic neuropathy |
Other organ manifestations |
Acute adrenal failure due to bilateral adrenal hemorrhagic infarction, osteonecrosis without histologic evidence for microvascular occlusion or vasculitis, acute sensorineural hearing loss |
Other coagulation abnormalities |
Thrombocytopenia, ITP, hypoprothrombinemia, protein S deficiency, acquired APC resistance, acquired inhibitors to specific coagulation factors, and AvWS |
Manifestations that are not included in consensus-based diagnostic criteria are italicized. |
|
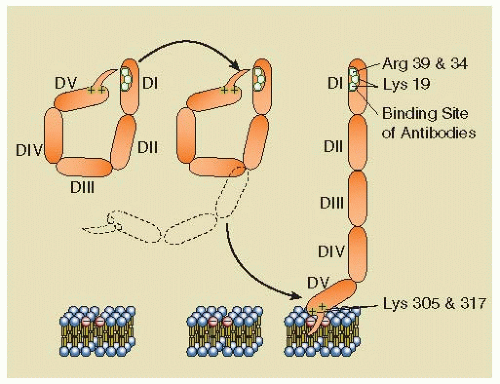
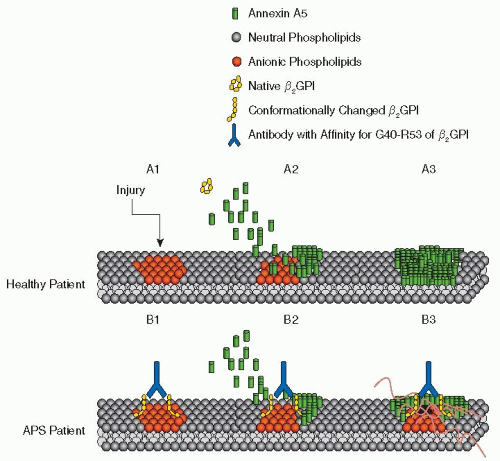

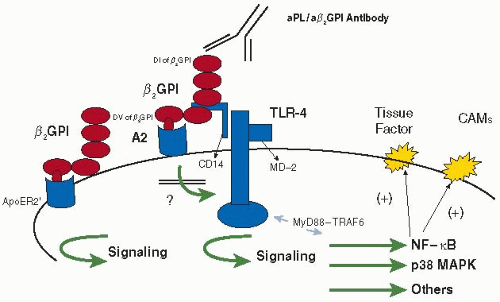
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Inherited Thrombocytopenias
Inherited Thrombocytopenias
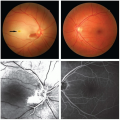 Unusual Sites of Arterial Occlusion
Unusual Sites of Arterial Occlusion
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient



