Thalidomide and Its Analogs in the Treatment of Hematologic Malignancies, Including Multiple Myeloma, and Solid Tumors
Thalidomide and Its Analogs in the Treatment of Hematologic Malignancies, Including Multiple Myeloma, and Solid Tumors
Jacob Laubach
Constantine S. Mitsiades
Teru Hideshima
Kenneth C. Anderson
Paul Richardson
Thalidomide was originally developed in the 1950s for the treatment of pregnancy-associated morning sickness. However, its extensive over-the-counter marketing in Europe was marked by the tragic consequences of teratogenecity,1 which triggered its subsequent withdrawal from the market.2,3 The teratogenic properties of thalidomide raised among oncologists the hypothesis that the potent inhibitory effects of this drug on growing fetal tissues, combined with the pathophysiologic similarities linking tumor biology and fetal development, might be redirected toward applications in cancer treatment.4 In fact, in the early 1960s at least two clinical trials of thalidomide for patients with advanced cancers were reported.5,6 In one of these trials, thalidomide was administered at daily doses of 300 to 2,000 mg in 71 patients with various types of cancers. No objective clinical responses were observed, except for resolution of a pulmonary metastasis in a patient with renal cell carcinoma (RCC).5 In the second trial, 21 patients with various types of advanced cancer (including two patients with multiple myeloma [MM]) received thalidomide at 600 to 2,000 mg daily doses, which led to palliation of symptoms in approximately one third of patients, while 2 patients had minimal slowing of their tumor’s growth.6 However, these results were not deemed sufficiently encouraging to warrant further clinical development efforts.
Thalidomide was therefore not pursued further as a potential anticancer drug for several decades. In the meantime, the drug gradually emerged as a therapeutic agent for a range of medical conditions, on the basis of anecdotal clinical evidence and converging research that suggested potential beneficial pharmacoimmunologic effects.7 Thalidomide gained renewed attention as an effective treatment of severe erythema nodosum leprosum (ENL),8,9 Behcet’s disease,10 graft-versus-host disease (GVHD),11 and oral ulcers and wasting associated with HIV infection.12,13 This reemergence of thalidomide was reflected by its Food and Drug Administration (FDA) approval in 1998 for the short-term treatment of cutaneous manifestations of moderate to severe ENL, together with its use as maintenance therapy to prevent recurrence of cutaneous ENL.7 This FDA approval was of critical importance for the clinical applications of thalidomide not only because the drug has since become a treatment of choice for ENL but also because the approval allowed for off-label uses of this medication in other disease states in which the immunomodulatory and antiangiogenic properties of thalidomide might be beneficial.7,14,15 To prevent any occurrence of teratogenic effects, thalidomide is now administered under strict guidelines to prevent fetal exposure to this medication.14
The interest in thalidomide as an anticancer drug was rekindled in the 1990s with the realization that tumor-associated vasculature is an important therapeutic target in a broad range of neoplasias and that thalidomide possesses substantial antiangiogenic properties in a wide range of in vivo and in vitro models of neovascularization.16, 17, 18, 19, 20, 21, 22, 23, 24 Indeed, thalidomide inhibited angiogenesis induced by basic fibroblast growth factor (bFGF) in the rabbit cornea micropocket assay or by vascular endothelial growth factor (VEGF) in a murine model of corneal vascularization.20,25 Based on these data from D’Amato, Folkman et al. as well as the fact that thalidomide is transformed to active metabolites with antiangiogenic activity in humans,26 thalidomide was evaluated for the treatment of various neoplasias.27, 28, 29, 30, 31 Of particular note was the well-chronicled decision to test thalidomide, at the suggestion of an especially enlightened wife of an MM patient, in a compassionate use study of three patients with advanced MM at the University of Arkansas. The encouraging evidence of clinical activity in two of these three patients led to a larger phase II effort,32 which confirmed the clinical activity of thalidomide against MM, and was followed by extensive clinical trials of thalidomide-based therapy for MM worldwide, as well as other applications in a broad range of other hematologic malignancies and solid tumors. Despite progress in its use and the fact that thalidomide is not associated with the classical pattern of toxicities seen with conventional DNA- or microtubule-targeting chemotherapeutics, this drug is not devoid of adverse effects. This realization led to development of thalidomide analogs that retain the clinical activity of thalidomide, without some of its prominent side effects.
In this chapter, we present a comprehensive review of the pharmacology of thalidomide and its analogs, a description of preclinical studies that illuminate the complex mechanisms of the action of thalidomide, and an overview of clinical trials involving thalidomide and its analogs in MM, other hematologic malignancies, solid tumors, and other cancer-related applications.
Mechanism of Action of Thalidomide and Analogs
Although it was originally hypothesized that the teratogenic and antitumor effects of thalidomide may have a common underlying mechanistic denominator, such as the antiangiogenic effect, the precise mechanisms responsible for the clinical activity of thalidomide have not been established. This can be attributed to a number of factors: (a) the preclinical in vitro and in vivo studies of thalidomide necessary to dissect its mechanisms of action are difficult to perform and interpret because of the enantiomeric interconversion and spontaneous cleavage of the drug to multiple metabolites in vivo,33 many of which have been incompletely characterized; (b) the in vivo activity of thalidomide likely requires metabolic activation by the liver, a finding that may account at least in part for the discordance between the modest, at best, activity of thalidomide in in vitro assays of antitumor activity26,34 and its potent in vivo effect; (c) a complex series of metabolic, immunologic, and antiangiogenic actions have emerged from preclinical studies; their possible relevance to anticancer therapy in humans is difficult to discern; (d) the chemical structure of thalidomide does not offer readily recognizable clues regarding possible intracellular molecular targets that might explain its clinical activity or profile of adverse events; and (e) the species-specific differences in the metabolism and other pharmacokinetic properties of thalidomide complicate the extrapolation of in vivo data from many animal models to the clinical setting in humans.25
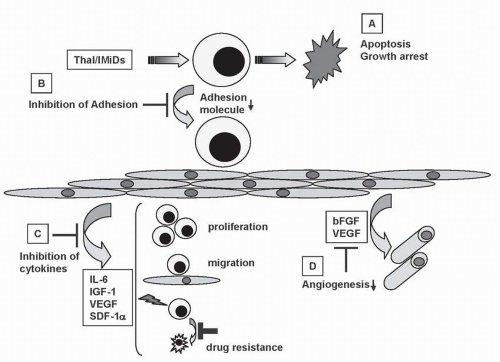
A wealth of mechanistic information has been acquired in MM, mainly because this is the disease setting in which thalidomide has demonstrated its most impressive clinical activity. Despite the modest activity of thalidomide in antitumor assays in vitro,35 this drug is currently considered to confer its in vivo anti-MM effects via at least four distinct, but potentially complementary, actions: (a) direct antiproliferative/proapoptotic antitumor effect,35 probably mediated by one or more of its in vivo metabolites36; (b) indirect targeting of tumor cells by abrogation of protective effects conferred to MM tumor cells by bone marrow stromal cells (BMSCs) via paracrine or autocrine secretion of cytokines and growth factor or via cell adhesion molecule mediated interactions35; (c) antiangiogenic effects; and (d) immunomodulatory effects, which contribute to enhanced antitumor immune response.37
Direct Antitumor Activity
The notion that thalidomide possesses direct antitumor effects in MM and other diseases is inferred from preclinical and clinical studies. Although the in vitro effect of thalidomide on proliferation and viability of MM cells is relatively modest,35 thalidomide derivatives, such as lenalidomide (Revlimid, CC-5013), and pomalidomoide (CC-4047, Actimid),35,36 have far more potent in vitro antiproliferative and proapoptotic properties than the parent compound. These effects were assayed in the absence of any other cell type (e.g., stromal, endothelial, or liver cells), which could facilitate either thalidomide metabolism or indirect effects on targets other than the tumor cells themselves.35,36 Therefore, the fact that at least some of the known in vivo thalidomide metabolites can have in vitro activity against tumor cells suggests that thalidomide may confer direct in vivo antiproliferative/proapoptotic effects via its metabolites. The precise mechanism(s) for this direct effect remain(s) under investigation. Cell cycle analyses, by propidium iodide staining, of thalidomide- and lenalidomide-treated MM cell lines indicate G0/G1 growth arrest, subsequently followed by increased sub-G1 peak, consistent with apoptosis and induction of cell death.35
Interestingly, clinically relevant concentrations of thalidomide derivatives, such as lenalidomide and pomalidomide, trigger suppression of the transcriptional activity of NF-κB in MM cells.36 NF-κB is well established as an important protective responder to DNA damage. It suppresses apoptosis by promoting the expression of intracellular antiapoptotic molecules, including the caspase inhibitors FLIP, XIAP, cIAP-2 or the antiapoptotic Bcl-2 family member A1/Bfl-1.38, 39, 40 Thalidomide thus may exert its in vivo effects, at least in part, by inhibition of NF-κB signaling in cells. It is also conceivable that because NF-κB protects cells from the proapoptotic effects of steroids or cytotoxic chemotherapeutics,36,38, 39, 40 the synergy of thalidomide and its analogs in concert with dexamethasone or cytotoxic chemotherapeutics may result from suppression of NF-κB.41, 42, 43, 44, 45, 46, 47, 48
Stroma-Tumor Interactions
Thalidomide and its derivatives modulate the adhesive interactions of MM cells with BMSCs.49 MM cells adhere to BMSCs and trigger their secretion of proliferative/antiapoptotic cytokines (e.g., interleukin [IL]-6).50, 51, 52 This event is mainly paracrine, is mediated by transcriptional activation of NF-kB in BMSCs,51 and dampens the sensitivity of MM cells to dexamethasone or cytotoxic chemotherapy.53 Thalidomide/IMiD blocks this MM-stromal paracrine interaction, significantly inhibits proliferation, and lessens drug resistance of MM cells in the BM microenvironment.
Inhibition of Cytokine Production and Antiangiogenic Action
Thalidomide also inhibits TNF-α production, while leaving the patient’s immune system otherwise intact.54 Thus, thalidomide is useful in various inflammatory disorders characterized by increased TNF-α secretion, such as ENL, mycobacterium tuberculosis infection, GVHD, and cancer- and HIV-related cachexia.
The precise mechanism mediating thalidomide-induced inhibition of TNF-α activity is not fully understood, but is apparently distinct from those of other TNF-α inhibitors, such as pentoxifylline and dexamethasone.55,56 Thalidomide may accelerate the degradation of TNF-α mRNA, thereby substantially (but not necessarily completely) suppressing the production of TNF-α protein.55,57 Interestingly, thalidomide can decrease the binding of the transcription factor NF-κB to its consensus DNA-binding sites, which include not only the actual TNF-α gene58 but also other genes modulated by TNF-α, in an NF-κB-dependent fashion.36 Even the antiangiogenic properties of thalidomide may be mediated, at least in part, by inhibition of TNF-α signaling, in view of the proangiogenic effects of TNF-α itself.20 However, the absence of a major effect of TNF-α in experimental models of angiogenesis and the inability of (at least some) potent TNFα inhibitors to directly influence angiogenesis suggest that thalidomide’s antiangiogenic effects cannot be attributed to TNF-α inhibition alone.20,25
Thalidomide and its analogs suppress the production of cytokines that regulate tumor cell proliferation and osteoclast function, including IL-6, IL-1β, IL-10, and TNF-α.59 Thalidomide also decreases the secretion of VEGF, IL-6,60 and bFGF by MM and/or BM stromal cells. These mechanisms are summarized in Figures 34-1 and 34-2. While characterizing the mechanism underlying the teratogenic effects of thalidomide, D’Amato et al. observed its antiangiogenic properties,20,25 which involved inhibition of the proangiogenic effects of β-FGF and/or VEGF.20,25,61 Further in vitro studies have suggested that the metabolites, and not the parent compound, are responsible for the antiangiogenic effect of thalidomide.62
FIGURE 34-1 Antitumor activity of Thal/IMiDs in the bone marrow (BM) milieu. Thal/IMiDs (A) induce G1 growth arrest and/or apoptosis in MM cell lines and patient cells resistant to conventional chemotherapy; (B) inhibit MM cell adhesion to BM stem cells (SCs); (C) decrease cytokine production and sequelae; (D) decrease angiogenesis, in the BM microenvironment.
Immunomodulation
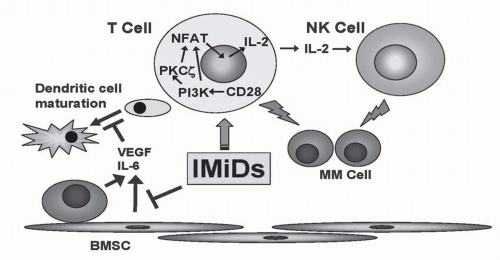
The precise effect of thalidomide on immune effector cells, for example, different subpopulations of lymphocytes, has not been consistent in studies published to date.63, 64, 65 Although thalidomide may not directly suppress lymphocyte proliferation,49 evidence suggests that it stimulates T-cell responses to tumor and may inhibit proliferation of already stimulated lymphocytes.64,66, 67, 68, 69 Thalidomide modifies the expression patterns of cell adhesion molecules on leukocytes, inhibits neutrophil chemotaxis, and inhibits TNF-α signaling and IL-12 production, enhances synthesis of IL-2, and inhibits IL-6.8,49,54,70, 71, 72 Of particular relevance to MM, thalidomide and its analogs augment natural killer (NK) cell-mediated cytotoxicity in MM.37 Thalidomide and IMiDs do not induce T-cell proliferation alone, but act as costimulators to trigger proliferation of anti-CD3-stimulated T cells from MM patients, accompanied by an increase in interferon-γ and IL-12 secretion. Importantly, treatment of patient peripheral blood mononuclear cells (PBMCs) with thalidomide or IMiDs triggered their lysis of autologous MM cells. Furthermore, in MM patients, thalidomide stimulated an increase in circulating CD3-CD56+ NK cells.37
FIGURE 34-2 Mechanisms of action of IMiDs in augmentation of host immune response. IMiDs augment differentiation of dendritic cells (DC) by inhibiting secretion of IL-6 and VEGF from multiple myeloma (MM) or bone marrow stem cells (BMSCs). IMiDs also stimulate natural killer (NK) cell activity by triggering IL-2 secretion from T-cells mediated by CD28/PI3-K/NF-AT2 (nuclear factor of activated T cells) signaling pathway. (Please see Color Insert.)
Pharmacology of Thalidomide
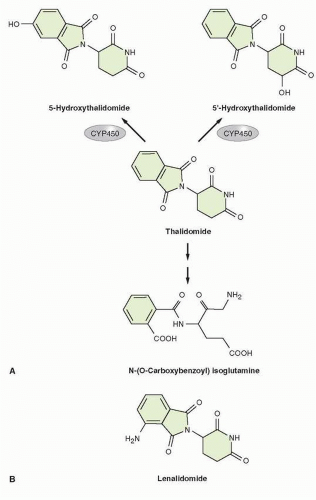
Thalidomide or α–N {phthalimido} glutarimide (C13O4N2H9) (gram molecular weight of 258.2)73 is a glutamic acid derivative, which contains two amide rings and a single chiral center (Fig. 34-3).73 The key features for thalidomine are shown in Table 34-1. The currently available formulation of thalidomide consists, at physiologic pH, of a nonpolar racemic mixture of S(−) and R(+) isomers, which are cell membrane permeable.73,74 The S isomer has been associated with the teratogenicity of thalidomide, while the R isomer has been linked with the sedative properties of the drug.25,74,75 Because of rapid interconversion of these two isomers at physiologic pH in vivo, efforts to generate formulations of only the R isomer have failed to neutralize the teratogenic potential of thalidomide.74,76
Pharmacokinetics
Pharmacokinetic studies of thalidomide in humans have been limited by the lack of suitable intravenous formulations due to its instability and poor water solubility. Therefore, the current knowledge on thalidomide pharmacokinetics is based on animal studies and on clinical trials in human patients receiving oral thalidomide.
FIGURE 34-3 (A) Proposed steps in metabolism of thalidomide (reference to be added). Thalidomide undergoes rapid, spontaneous nonenzymatic hydrolytic cleavage at physiologic pH, with generation of up to 50 metabolites. (B) Chemical structure of lenalidomide.
As shown in Table 34-2, the pharmacokinetics properties of thalidomide are variable, with the t½ of parent compounds in plasma falling in the range of 4 to 9 hours.34
Absorption
Oral administration of thalidomide at a dose of 100 mg/kg in animals led to peak serum concentrations within 4 hours77; the bioavailability was independent of dose. Subsequent studies in humans demonstrated a similar pharmacokinetic pattern, with approximately 4-hour mean time to reach peak concentration (tmax) with a thalidomide dose of 200 mg.23, 24, 25,29 In particular, studies on single-dose thalidomide have been conducted in healthy volunteers, patients with HIV infection, and patients with hormone refractory prostate cancer (see Table 34-2); the time to peak concentration in plasma ranged from 3 to 6 hours, suggesting slow absorption from the gastrointestinal tract (GI).78, 79, 80 While the area under the curve (AUC) correlates with the thalidomide dose, the maximum concentration (Cmax) is highly variable, which further reflects the variability of GI absorption.81,82 Correction for ideal body weight or body surface area does not lessen the variability.78,79 This variability has further confounded efforts to delineate the pharmacokinetic properties of thalidomide in humans and define a dose-response relationship in therapeutic trials.
TABLE 34.1Key features of thalidomide and lenalidomide
Thalidomide
Lenalidomide
Classification
Glutamic acid derivative with two amide rings and a single chiral center
Thalidomide analog with modification through elimination of carbonyl group and addition of amine
Direct antitumor activity via induction of apoptosis Blockade of tumor cell-stroma interactions Inhibition of cytokine production, including TNF-α and VEGF Immune-modulating, with stimulation of T-cell and NK cell activity
Mechanisms of action similar to thalidomide, but overall higher potency compared to thalidomide, in preclinical most assays reported in the literature
Clinical pharmacology
Oral bioavailability: Rapid absorption with time to peak plasma concentration at 4 h
Pharmacokinetic parameters: Rapid absorption with peak plasma concentration at 0.6-1.5 h
Pharmacokinetic parameters: Pharmacokinetic data limited by lack of intravenous formulation due to instability and poor water solubility Distribution: Widely distributed throughout most tissues and organs without significant binding by plasma proteins t½ life: 4.1-18.3 h Metabolism: Rapid, spontaneous nonenzymatic hydrolytic cleavage. Limited metabolism via CYP450 system. Elimination: Primarily through metabolism
Pharmacokinetic parameters: Pharmacokinetic data limited by lack of intravenous formulation due to instability and poor water solubility. Linear pharmacokinetics with plasma exposure proportional to dose. t½ life: 3-4 h. With moderate to severe renal impairment, 9 h. Metabolism: Not a substrate of hepatic metabolic enzymes; unchanged lenalidomide is predominant circulating compound. Elimination: 65%-85% eliminated through urinary excretion of unchanged drug
FDA indications
Newly diagnosed and relapsed/refractory multiple myeloma Cutaneous erythema nodosum leprosum
Relapsed/refractory multiple myeloma Low-risk or intermediate-1-risk myelodysplastic syndrome associated with 5q cytogenetic abnormality
Dosage
100-200 mg once daily
Multiple myeloma: 25 mg daily days 1-21 of 28-d cycle Myelodysplastic syndrome: 10 mg once daily
Drug interactions
The toxicity of anti-TNF agents such as abatacept, anakinra, canakinumab, and certolizumab may be enhanced by thalidomide.
The toxicity of anti-TNF agents such as abatacept, anakinra, canakinumab, and certolizumab may be enhanced by lenalidomide.
Thalidomide may enhance the myelosuppressive effects of leflunomide and trastuzumab.
Lenalidomide may enhance the myelosuppressive effects of leflunomide and trastuzumab.
Thalidomide is a known teratogen and pregnancy should be avoided throughout therapy. Thalidomide should be used with caution in patients at risk for venous thromboembolic disease. Neuropathy can be permanent. Caution is advised when administering to patients with history of cardiovascular disease.
As an analog of thalidomide, lenalidomide has teratogenic potential. Pregnancy should be avoided while taking lenalidomide. Formulations may contain lactose, and caution should be exercised when administering to patients with lactose intolerance.
TABLE 34.2Single-dose pharmacokinetic parameters of thalidomide in humans
Source: Hideshima T, et al. Thalidomide and its analogs overcome drug resistance of human multiple myeloma cells to conventional therapy. Blood 2000;96:2943-2950.
Distribution
In animal studies, thalidomide is widely distributed throughout most tissues and organs,77 without significant drug binding by plasma proteins.79,80 Thalidomide is detected in semen of rabbits following oral administration73,83 and has been shown to be present in semen of patients after a period of 4 weeks of therapy, with levels that correlate with serum levels.84
Metabolism
Thalidomide undergoes rapid and spontaneous nonenzymatic hydrolytic cleavage at physiologic pH (Fig. 34-3), generating up to 50 metabolites, 5 of which are considered to be primary metabolites.26,74,76,77,85 The majority of these metabolites are unstable and their rapid degradation under physiologic conditions has complicated their characterization.86 Although in vitro studies in rat cells suggested that thalidomide induces cytochrome (CYP) 450 isoenzymes, subsequent evaluation of single- or multiple-dose pharmacokinetic parameters of oral thalidomide at 200 mg daily in healthy volunteers showed that thalidomide does not inhibit or induce its own metabolism over a 21-day period in humans. It is therefore considered that only limited metabolism of thalidomide occurs via the hepatic CYP 450 system.87
Only gold members can continue reading. Log In or Register to continue
May 27, 2016 | Posted by drzezo in ONCOLOGY | Comments Off on Thalidomide and Its Analogs in the Treatment of Hematologic Malignancies, Including Multiple Myeloma, and Solid Tumors