Disorders associated with:
Thrombocytopenia
Platelet dysfunction (qualitative platelet disorders)
Coagulation disorders
Autosomal recessive
Autosomal recessive
X-linked
Bernard-Soulier syndrome
Bernard-Soulier syndrome
Factor VIII deficiency (hemophilia A)
Congenital amegakaryocytic thrombocytopenia
Glanzmann’s thrombasthenia
Factor IX deficiency (hemophilia B)
Storage pool disorder
Gray platelet syndrome
Gray platelet syndrome
Autosomal recessive
Thrombocytopenia with absent radius
Cyclooxygenase deficiency
Factor XI deficiency
Thromboxane synthase deficiency
Prothrombin deficiency
Factor V deficiency
Autosomal dominant
Factor VII deficiency
May-Hegglin anomaly
Scott’s syndrome
Factor X deficiency
Quebec platelet disorder
Factor XIII deficiency
Epstein’s syndrome
Afibrinogenemia
Fechtner’s syndrome
Hypofibrinogenemia
Sebastian’s syndrome
αz Antiplasmin deficiency
Montreal platelet syndrome
Plasminogen activator inhibitor deficiency
Heredity macrothrombocytopenia
Combined deficiency of factors V/VIII
X-linked
Autosomal dominant
Wiskott-Aldrich syndrome
von Willebrand’s disease
X-linked thrombocytopenia
Dysfibrinogenemia
GATA-1 mutation
6.1 Qualitative Platelet Disorders
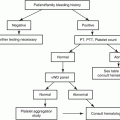
In these disorders, platelet dysfunction is associated with a normal platelet count. Normal platelet function results from several platelet activities. Platelet adhesion to subendothelium is the initial response to vascular injury, and is mediated by the plasma adhesion protein, von Willebrand’s factor (vWF) and its platelet receptor, glycoprotein (GP) 1b. Platelet-platelet interaction (aggregation) is mediated by plasma fibrinogen and its platelet receptor, GP IIb-IIIa. Following platelet activation, secreted ADP amplifies platelet aggregation to generate the platelet thrombus [1]. These hemostatic events are summarized in Fig. 6.1. The platelet plug is consolidated (reinforced) by fibrin generated from the coagulation mechanism. All of these platelet functions (adhesion, aggregation, secretion) can be measured in the laboratory, and deficient platelet GP function can be inferred or quantitated. Screening tests for platelet function such as the bleeding time are not recommended and the utility of other screening tests such as the PFA-100 is unproven (see Chaps. 7 and 3).
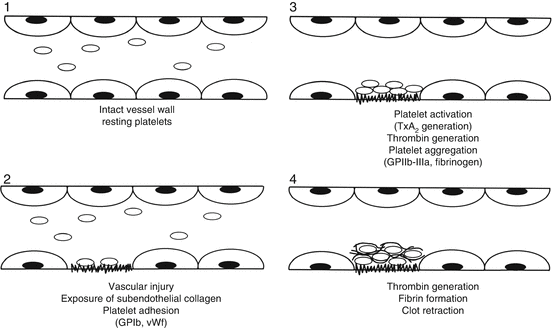
Fig. 6.1
Sequence of hemostatic events following vascular injury. 1 In the resting state, platelets are non-activated and the endothelium is intact. 2 Following vascular injury, subendothelial components are exposed to induce platelet adhesion, mediated by vWF. 3 Collagen exposure activates platelets leading to platelet secretion, thromboxane A2 generation and formation of thrombin. These events lead to platelet aggregation. 4 Thrombin generation results in cross-linked fibrin that reinforces the platelet plug (From Rodgers [1], p. 306, with permission)
6.2 Platelet Aggregation
The traditional assay for platelet function is platelet aggregation using platelet-rich plasma. Since countless prescription and over-the-counter medications and dietary supplements may impair platelet function, patients’ medication lists must be reviewed by the coagulation laboratory prior to testing. Drugs containing aspirin must be discontinued 10–14 days prior to phlebotomy, while most other drugs should be stopped 4–7 days before phlebotomy. Citrated blood is obtained from the patient and a control subject (who also is taking no interfering medications), and platelet-rich plasma (PRP) is prepared by slow-speed centrifugation. The blood sample should not be refrigerated. The PRP specimens are mixed with patient platelet-poor plasma to obtain a final platelet count ~250,000/μL [2].
Platelet aggregation is based on spectrophotometric monitoring of the turbid PRP sample after addition of various agonists (platelet activating stimuli). Agonist-induced platelet aggregation results in decreased turbidity and increased light transmittance. Figure 6.2 illustrates normal platelet aggregation patterns with PRP. The agonists used in platelet aggregation studies commonly include ADP (low-dose 0.5–2 μg/mL; high-dose 10–20 μg/mL), dilutions of a collagen suspension, arachidonic acid, and ristocetin (low-dose 0.5 μg/mL; high-dose 1–2 μg/mL). Some laboratories also use epinephrine as an agonist. This panel of agonists will test various platelet functions, including adhesion, aggregation, secretion and activation [2].
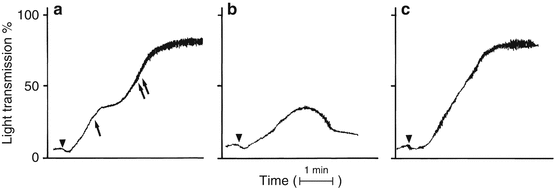
Fig. 6.2
Normal platelet aggregation patterns. (a) The primary and secondary waves of the platelet aggregation response. The primary wave (single arrow) results from agonist-induced platelet aggregation whereas the secondary wave (two arrows) results from platelet secretion of endogenous ADP and recruitment of additional activated platelets. (b) Low-dose agonist-induced primary wave aggregation. The amount of stimulus is insufficient to fully activate the platelet and induce secretion of stored ADP. (c) High-dose agonist-induced platelet aggregation in which the primary and secondary aggregation waves have merged into a single aggregation wave. Triangles indicate addition of the agonist (From Rodgers [12], with permission)
Zhou and Schmaier have published platelet aggregation procedures to standardize performance and interpretation of platelet function testing [3]. They ask patients to avoid all medications for 10 days prior to assay, to avoid coffee on the day of testing, and to fast for at least 4 hours before phlebotomy.
These authors recommend that each aggregation run last for 5–10 minutes, to allow observation of both primary and secondary aggregation waves, as well as the phenomenon of platelet disaggregation which may occur in some patients with platelet dysfunction.
Interpretation of platelet aggregation patterns can be done in two ways: calculation of the extent (percent) of aggregation, or determination of the initial rate of aggregation per minute. Zhou and Schmaier have published normal values for platelet aggregation results; most agonists result in maximal extent (percent) of aggregation of 70–80 % [3]. Recommendations from a committee of the ISTH on standardization of light transmission aggregometry summarize pre-analytical variables, blood collection techniques, platelet preparation methods, agonist choice and concentration and data reporting [4].
Figure 6.3 summarizes platelet aggregation profile results in patients with Glanzmann’s thrombasthenia, Bernard-Soulier syndrome, and storage pool/secretion disorders (thrombopathies). Glanzmann’s thrombasthenia platelets have deficient GPIIb-IIIa receptors, so that aggregation dependent on this receptor is decreased or absent. Consequently, aggregation induced by ADP, collagen, arachidonic acid (all agonists except ristocetin) is abnormal. In contrast, patients with Bernard-Soulier syndrome have platelets deficient in GP1b receptors, so that aggregation dependent on this receptor is decreased or absent. Since ristocetin is the only agonist dependent on normal GP1b function, Bernard-Soulier syndrome platelets have normal aggregation responses to all agonists but ristocetin [2].
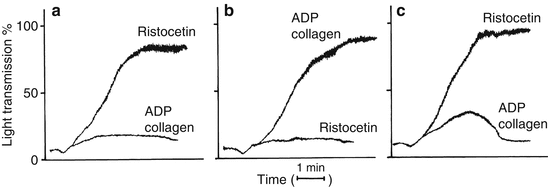
Fig. 6.3
Platelet aggregation profiles in vWD and the inherited qualitative platelet disorders. Typical responses to ADP, collagen, and ristocetin are shown. Three patterns of aggregation are shown: (a) Glanzmann’s thrombasthenia. (b) Bernard-Soulier syndrome and vWD. Both diseases give similar aggregation results with decreased or absent response to ristocetin. The two diseases are distinguished by Bernard-Soulier patients having giant platelet morphology with thrombocytopenia and vWD patients having normal platelet morphology and decreased ristocetin cofactor activity. (c) Thrombopathies (secretion defects and abnormalities in prostaglandin synthesis) (From Rodgers [12], with permission)
Platelets from patients with storage pool disorder or secretion defects will exhibit normal primary aggregation responses to all agonists, but since the defect is in platelet activation and/or secretion, secondary aggregation is defective to all agonists except ristocetin [2].
Platelet aggregation results can be reported quantitatively (percentage change of transmittance) or qualitatively (normal or abnormal), with a comment as to the type of abnormality and possible diagnosis. It is important to remember that numerous common medications can induce marked platelet dysfunction, and this potential confounding issue should be considered when interpreting an abnormal platelet aggregation study.
Table 6.2 summarizes the platelet aggregation profiles seen with the inherited qualitative platelet disorders. Although von Willebrand’s disease (vWD) is best diagnosed with other tests, platelet aggregation can be used to diagnose vWD. Deficiency of vWF results in a profile where all agonist responses are normal, except for ristocetin, a pattern similar to that seen in Bernard-Soulier syndrome. In Bernard-Soulier syndrome, the defect is in the vWF receptor (GP1b); in vWD, the defect is absent vWF. The two disorders can be distinguished by assay of vWF activity with the ristocetin cofactor assay; levels are normal in Bernard Soulier syndrome and decreased in vWD (Table 6.2).
Table 6.2
Platelet aggregation profiles in the inherited qualitative platelet disorders and von Willebrand’s disease
Aggregation response | |||||
|---|---|---|---|---|---|
ADP | Collagen | Ristocetin | Comments | ||
Disorders | Primary | Secondary | |||
Bernard-Soulier syndrome | N | N | N | A | Giant platelets and thrombocytopenia are present. vWF activity assay is normal |
Glanzmann’s thrombasthenia | A | A | A | N | Normal platelet morphology |
Storage pool disorder | N | A | N | N | Platelet morphology by light microscopy is usually normal. Electron microcopy is abnormal |
Secretion defect | N | A | N | N | Platelet morphology by light and electron microscopy is normal |
vWD | N | N | N | A | Platelet morphology is normal. vWF activity assay is decreased |
The key clinical distinction in evaluating patients for qualitative platelet disorders is to differentiate bleeding from a platelet disorder vs. vWD. Both disorders have similar clinical features, but are treated differently – vWD patients should receive a vWF replacement product, while patients with platelet dysfunction should receive platelet transfusions for significant bleeding.
Whole blood aggregation methods are also available that are less laborious than the PRP method. Certain whole blood assays also report platelet secretion data in addition to aggregation data, permitting direct identification of platelet dysfunction due to secretion defects.
6.3 von Willebrand’s Disease
vWD is the most common inherited bleeding disorder, affecting ~1 % of the population. The disorder is characterized by deficiency or qualitative abnormality of vWF, the plasma adhesion protein necessary for primary hemostasis. vWF circulates in the blood in a complex with factor VIII, the plasma protein deficient in hemophilia A. In vivo, vascular injury results in exposure of subendothelial components that promote vWF-mediated platelet adhesion. This event is dependent primarily on the highest molecular weight component (multimers) of vWF. In vitro assays of platelet function using ristocetin mimic in vivo platelet adhesion; such assays (ristocetin-induced platelet aggregation, ristocetin cofactor activity) measure vWF function [2].
6.4 Assays for vWF
Several assays for vWF are available to assess patients with bleeding disorders for vWD. Most commonly, a vWD “panel” is obtained that measures antigenic and functional properties of the factor VIII – vWF complex. Panel components include vWF antigen, a quantitative assay of vWF, as measured by ELISA, Laurell immunoelectrophoresis, or immunoturbidimetric assay; vWF activity, as measured by a test called ristocetin cofactor activity; and factor VIII activity, as measured by coagulation or chromogenic substrate assay. Of these three panel components, ristocetin cofactor activity is frequently the most abnormal, but assaying all 3 components is recommended. If an abnormal vWD panel result is obtained, the patient’s disorder is further classified by multimeric analysis, in which the patient’s vWF is analyzed by immunoblot and characterized as type 1 (quantitative mild to moderate deficiency), type 2 [qualitative abnormality in which high-molecular-weight multimers are absent (type 2B), or high- and intermediate-molecular-weight multimers are absent (type 2A)], or type 3 (absence of vWF). This classification is clinically important since patients with type 1 vWD, but not types 2 or 3 vWD may respond to desmopressin, a non-transfusion therapy that can increase plasma vWF levels. Another assay that may be useful in diagnosing patients with vWD when the standard panel results are normal is ristocetin-induced platelet aggregation [2].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


