Chapter 53 Testicular Cancer
Testicular tumors are uncommon in the general population but are some of the most common and important malignant tumors in young men. Worldwide, 52,000 new cases and 9,000 deaths were estimated from this disease in 2008.1 The vast majority are primary germ cell tumors (GCTs); the others include lymphomas and sarcomas. The incidence of GCTs has doubled in the past 30 years. Although most patients (70% to 80%) present with early-stage, highly curable disease, the continued rise in incidence of these tumors presents a major challenge. Management is based on histologic type and disease extent. Advances in chemotherapy, imaging, and multidisciplinary care have led to improvements in outcomes over the past three decades. In this chapter, we will discuss the management principles for patients with primary GCTs of the testis, malignant extragonadal GCTs, and other rare testicular tumors.
Etiology and Epidemiology
Testicular cancer accounts for only 1% to 2 % of all cancers in men in most populations across the world.2 Germ cell testicular tumors are the most common solid malignant tumors in men between 20 and 35 years of age, and it was estimated that in the United States in 2010, there would be 8480 new cases and 350 deaths from testicular cancer.3 The cumulative lifetime risk of developing a GCT for a white man in the United States is 0.2%.4 Although the disease is rare, the observed rising incidence is a concern. Between 1973 and 2003 the incidence of GCTs rose by 61% in the United States, with the major rise occurring in seminomas rather than nonseminomas.5
Risk factors for testicular germ cell cancer (TGCC) include a family history of cancer, the presence of an undescended testis or gonadal dysgenesis, subfertility, and testicular microlithiasis. Men with a history of cryptorchidism have an approximately six-fold increased chance of developing testicular cancer.6 Orchiopexy prior to puberty appears to lower the risk of developing a subsequent tumor and may help preserve Leydig cell function and enhance fertility.7,8 Although most testicular cancers in men with a history of maldescent occur on the ipsilateral side, approximately 5% to 20% develop in the contralateral testicle.6 The mechanism by which cryptorchidism increases the risk of developing GCTs is unknown, but the effects of maldescent on the testis (i.e., increased temperature or increased risk of trauma if the testis is in the inguinal region) have been suggested as possible factors. The increase in the incidence of tumors in the contralateral testicle, however, suggests that maldescent and testicular cancer may result from the same prenatal etiologic process. Other genitourinary abnormalities associated with testicular cancers include hydrocele and hypospadias.9
There is considerable geographic and ethnic variation in the incidence of testicular tumors, with the highest incidence being reported from Denmark (8.4 per 100,000 men per year) and Switzerland (6.2 to 8.8 per 100,000 men per year).8 The incidence of testicular cancer is lower in non-Europeans as compared with Europeans. In the United States, white men (6.36 per 100,000 per year) are five to six times more likely to develop testicular cancers than black men (1.30 per 100,000 per year); low incidence rates are seen in other ethnic groups, such as Americans of Chinese and Japanese descent.5 A high incidence of testicular cancer is seen in some nonwhite populations such as the Maoris in New Zealand and Native Americans.10
Prior testicular cancer is a major risk factor for the development of a contralateral malignant tumor. In a large population-based follow-up study of 29,515 patients with unilateral testicular cancer, the cumulative risk (at 15 years after diagnosis of the primary tumor) of developing a contralateral malignant tumor was 1.9%.11
Heritability of Testicular Germ Cell Cancer
Given the strong familial association of TGCC, extended pedigrees with this disease are curiously rare. Most families with TGCC have two affected members, most commonly sibling pairs. The International Testicular Germ Cell Linkage Consortium has carried out two large-scale genome scans, neither of which provided convincing or replicable evidence for susceptibility gene(s).12 Candidate association studies have demonstrated that rare gr/gr deletions of the Y chromosome predispose to TGCC, but such deletions cannot explain more than a fraction of heritable cases of this disease.
More recently, two groups carried out association studies, based on genome-wide, single-nucleotide polymorphisms (SNPs), of patients with TGCC and controls.13,14 Both research groups identified strong evidence for two susceptibility loci for seminoma and nonseminoma; one locus is on chromosome 12p22, within the KITLG gene, encoding the ligand for the receptor tyrosine kinase KIT, and the second locus is on chromosome 5q31.3 near the SPRY4 gene, encoding sprouty 4, an inhibitor of the MAP kinase pathway. A third locus on chromosome 6 was identified by only one of the research groups and is not associated with a known gene. The relative risk was approximately 3 for the KITLG locus and about 1.4 for the other two loci. Both the KITLG gene and the SPTY4 gene are biologically feasible candidates because their products lie in the same signal transduction pathway. Moreover, activating mutations of the KIT gene have been identified in a subset of seminomas, while paradoxically, deletions of the KIT gene predispose to germ cell cancer in the SV/129 mouse strain. Of note, cases in the association studies included patients with and patients without a family history of TGCC, but none of these loci were enriched in the familial cases and neither were they associated with other abnormalities such as an undescended testis.
As mentioned above, the SV/129 mouse strain is particularly susceptible to the development of germ cell cancer, and this system has been exploited by Nadeau and colleagues14a to map several genes predisposing to the disease. Curiously, none of the corresponding loci in humans appear to have any role in TGCC development.
Environmental Factors and Testicular Germ Cell Cancer
Other factors linked to the development of TGCCs include a history of testicular trauma, an increased body mass index, immunosuppression following organ transplantation, and human immunodeficiency virus (HIV) infection.2,15–17 There is no evidence to suggest a causal relationship between testicular trauma and the development of a tumor, and the likely explanation is that testicular trauma leads to examination of the testes. Because of the age distribution of testicular cancer, exposure must occur early in life if there is an environmental contribution to the etiology of these tumors. Prenatal factors linked to the later development of testicular cancers include threatened miscarriage, excessive maternal nausea, and birth by caesarean delivery.2 To explain these associations it has been suggested that exposure of the germinal epithelium in utero to an elevated level of free unbound maternal estrogen could give rise to subsequent cryptorchidism and an elevated risk of developing a testicular tumor; the prenatal estrogen theory remains unproven, however.2
Prevention and Early Detection
The identification of carcinoma in situ (CIS) or testicular intraepithelial neoplasia (TIN) as a precursor of testicular GCTs has raised the possibility that the development of invasive testicular cancer could be prevented by treating CIS. In adults, CIS is found adjacent to GCTs in virtually 100% of cases, and it is thought that, with the exception of spermatocytes seminoma, CIS precedes the development of all invasive tumors.18 The natural history of testicular CIS is unknown, but the Danish experience suggests that all cases of adult CIS will ultimately progress to invasive cancer.18
The diagnosis of testicular CIS can only be made by testicular biopsy. Because the incidence of CIS in the general population is low (at most, 0.7%), screening biopsies are currently not recommended. They should be considered in high-risk patients, however, including those with presumed extragonadal germ cell cancer, intersex individuals, and select patients with contralateral GCT (age <40 years and testicular volumes of <12 mL).19
The management of patients with testicular CIS is controversial. Orchiectomy has been suggested for unilateral disease, and in cases diagnosed after orchiectomy for GCT, three options can be considered and discussed with the patient: orchiectomy, low-dose RT, or surveillance.19 Although both orchiectomy and RT offer definitive treatment for testicular CIS, they both destroy any residual fertility. Surveillance, with careful follow-up of the affected testis, is a reasonable option, especially given the excellent prognosis for metachronous testicular cancers.20
Biologic Characteristics and Molecular Biology
GCTs have a distinctive capacity for totipotential differentiation as demonstrated by the frequent finding of combinations of choriocarcinoma, embryonal carcinoma, and seminoma in a single tumor. They also can retain their ability to differentiate as displayed by the not infrequent identification of mature teratoma in residual post-treatment retroperitoneal masses. Cytogenetic analysis of GCTs has shown that chromosome numbers are more homogenous in seminomas than in nonseminomas.21–23 Triploid and tetraploid chromosomal patterns are common in seminomas, and hyperdiploid to hypertriploid counts are common in nonseminomas. A characteristic chromosome anomaly in GCTs of all histologic types is the presence of an isochromosome of the short arm of chromosome 12.24 This isochromosome, first reported by Atkin and Baker in 1982,21 consists essentially of two chromosome 12 short arms. It is present in more than 80% of cases, and GCTs without a 12p isochromosome have extra copies of 12p segments incorporated into other chromosomes. The 12p isochromosome is also found in testicular CIS.24 Isochromosome copies tend to be more numerous in nonseminomas than in seminomas.
It is not known how these chromosomal changes contribute to the development of the neoplastic phenotype. The frequent finding of the 12p isochromosome in both CIS and invasive tumors, however, indicates that this chromosome plays an important role in the biology of these tumors. The possibility of amplification of normal or modified genes, such as the DDX1 gene on the 12p isochromosome, is currently being investigated.25 Proto-oncogenes present on the short arm of chromosome 12 could be activated by point mutations, deletions, or translocations to become oncogenes, which could then act in a dominant manner.26 The KRAS gene is located on the short arm of chromosome 12, and amplification or enhanced expression of this gene has been reported to occur in testicular tumors and derived cell lines. Other candidate genes being studied include the c-KIT oncogene and its ligand KITGL or SCF, the c-MOS oncogene, and the CCND2 gene. In addition to chromosome 12, 17q is overrepresented in 50% of cases of GCT; this area contains a number of genes of interest, including the GRB7 gene and the plakoglobin gene.22
The study of the genetics and molecular biology of GCTs has enhanced our understanding of these tumors. To date, however, cytogenetics has had little impact in the clinic, with the possible exception of the use of various markers for the identification of non-TGCCs located elsewhere in the body.27–29
Pathology and Pathways of Spread
Pathology
Testicular cancers can arise from intratesticular and paratesticular cells (Table 53-1). The vast majority are of germ cell origin, and three major classification schemes have been in use worldwide30–32 (Table 53-2). The Dixon and Moore classification as modified by Mostofi has been adopted by the World Health Organization (WHO) and is the classification scheme most widely used in North America.33
TABLE 53-1 Histologic Classification of Testicular Neoplasms
TABLE 53-2 Three Classifications of Germ Cell Tumors
| Dixon and Moore, 195330 | WHO Classification33 | Pugh, 197632 |
|---|---|---|
For clinical purposes, GCTs are classified into two major groups: seminomas and nonseminomas (NSGCTs). It should be noted that patients with pure seminoma may have recurrence with pure NSGCT, and vice versa. Approximately 60% of GCTs are pure seminomas, 30% are NSGCTs, and 10% are mixed tumors (both seminomatous and NSGCT elements are present).34 Patients with mixed tumors are clinically considered to have NSGCT, the only exception being tumors with syncytiotrophoblastic cells in cases of seminoma.
Carcinoma in Situ
Intratubular germ cell neoplasia, or CIS, is felt to precede the development of all cases of seminoma and NSGCT in adults (with the exception of spermatocytic seminoma).35 On light microscopy, CIS cells closely resemble seminoma cells and in most cases are found within the seminiferous tubules. Cytologically, there is no difference between the CIS cells that develop into seminomas and those that develop into NSGCTs. In the general population, the incidence of CIS is very low (0.2%), but it is somewhat higher in men with impaired fertility (0.5%) and in those with cryptorchid testes (2% to 4%).18
Seminomas
Seminoma, the most common type of testicular GCT, is most often seen in the fourth decade of life. On gross examination, the tumors are usually well demarcated from the residual testicular tissue and rarely have foci of necrosis or hemorrhage. On microscopic examination, the classical or typical seminoma type is made up of large cells with abundant cytoplasm divided by connective tissue septae into sheets or cords.36 These cells typically have round, hyperchromatic or vesicular nuclei with prominent nucleoli. Frequently, there is a lymphocytic infiltrate, and macrophages, plasma cells, and multinucleated giant cells are often present. Syncytiotrophoblasts are present in 15% to 20% of cases, and their presence does not appear to alter the prognosis.
On immunohistochemical testing, virtually all seminomas express placental leukocyte alkaline phosphatase (PLAP) and do not express low-molecular-weight keratins, blood group antigens, or vimentin. Several histologic variants of seminoma have been identified, including anaplastic seminoma and spermatocytic seminoma. Anaplastic seminoma is diagnosed when there are three or more mitoses seen per high-power field.36 Spermatocytic seminoma is a rare subtype mainly seen in older men and is not associated with CIS or bilateral disease.35 Also, these tumors do not stain for PLAP on immunohistochemical testing and rarely, if ever, metastasize. An atypical variant of seminoma with some features similar to those of NSGCT on immunohistochemical examination has been reported, although its morphologic appearance is similar to that of classical seminoma.37 Usually, there is little or no lymphocytic infiltrate and the tumor cells have less cytoplasm than classical seminoma cells. In terms of prognosis, this atypical variant appears to have the same prognosis as classical seminoma.
Nonseminomatous Germ Cell Tumors
Nonseminomatous tumors make up 40% of TGCCs and occur most commonly in the third decade of life. In the WHO classification system, NSGCTs include embryonal carcinoma, teratoma (mature, immature, or with malignant differentiation), choriocarcinoma, yolk sac tumor, and mixed GCTs. Most tumors are mixed, with two or more cell types present. Although some tumors have a component of seminoma, the association of seminoma within a histologically confirmed NSGCT has no major impact on the clinical outcome.38 Patients with combined tumors present at an age (median, 33 years) intermediate between those with seminoma (median, 36 years) and those with nonseminoma (median, 27 years).38 On gross examination, there is usually a soft irregular mass poorly demarcated from the surrounding testicular tissue, and a considerable amount of necrosis and hemorrhage is often present. Immunohistochemical studies usually demonstrate cytoplasmic expression of low-molecular-weight keratins in embryonal carcinomas, and yolk sac elements, low-molecular-weight keratin, and/or vimentin expression in mature teratomas.
Pathways of Spread
Lymphatic spread is the most common route of metastatic spread. The lymphatic drainage of the testis is directly to the para-aortic lymph nodes. There are differences in the distribution of metastases from left or right testicular tumors. The left testicular vein drains to the left renal vein, and the lymphatic drainage is primarily to the lymph nodes in the para-aortic area, directly below the left renal hilum. On the right side, the testicular vein drains directly to the inferior vena cava below the level of the renal vein, and, therefore, paracaval and interaortocaval nodes are the first ones to be involved in right-sided tumors. Figure 53-1 shows the distribution of retroperitoneal lymph node metastases in NSGCT.39 Contralateral nodal involvement occurs in approximately 15% of cases and is rarely found in the absence of ipsilateral involvement. Supradiaphragmatic spread can occur through the thoracic duct, and although left supraclavicular nodal disease is infrequent at presentation, it is often seen at the time of relapse.
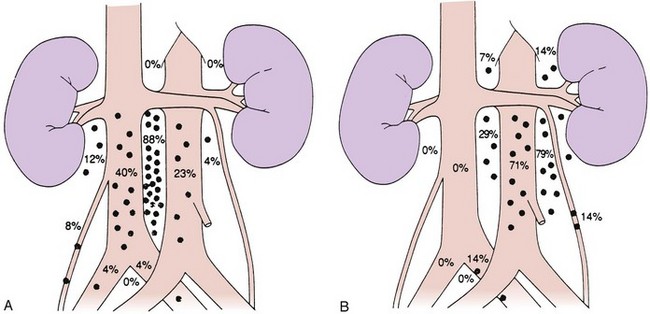
Pelvic and inguinal lymph node involvement is rare (<3%). Factors predisposing to inguinal lymph node involvement include prior scrotal or inguinal surgery, scrotal orchiectomy with incision of the tunica albuginea, tumor invasion of the tunica vaginalis or lower third of the epididymis, and cryptorchid testis.40,41 Disruption of lymphatic vessels in the spermatic cord during inguinal surgery has been shown to induce anastomoses between the testicular lymphatic vessels and the regional lymphatics destined for inguinal or pelvic lymph nodes. In an occasional patient, a connection with the contralateral inguinal lymph nodes may be established, but this is very uncommon. In a small proportion of patients with inguinal relapse, no predisposing factors may be apparent.40
In patients with NSGCTs, hematogenous spread occurs early in the course of the disease. The lung parenchyma is the commonest site of hematogenous spread, but liver, bone, brain, kidney, and gastrointestinal metastases are also seen. In a review of over 5000 patients with metastatic GCT, pulmonary metastases were present in 44% of cases and liver metastases in 6% of cases, with all other areas of hematogenous spread present in 1% or less of cases.42 Mediastinal and neck node involvement was present in 11% to 12% of cases.
Clinical Manifestations, Patient Evaluation, and Staging
Clinical Manifestations
Patients with testicular tumors most commonly present with a painless testicular mass. Up to 45% of patients may have testicular pain, with signs and symptoms suggestive of acute epididymitis in up to 25% of patients. Much less common presenting features include those related to the presence of metastases, for example, back pain, dyspnea, and gynecomastia (from malignant tumors that produce HCG). Up to 3.5% of patients—those with very high HCG levels—may develop hyperthyroidism because the alpha subunit of HCG is identical to that of thyroid-stimulating hormone (TSH) and may therefore stimulate the thyroid.43 The differential diagnosis of a testicular mass (in addition to tumor) includes torsion, hydrocele, varicocele, spermatocele, and epididymitis. A small percentage of tumors are associated with a hydrocele, so the presence of transillumination on examination does not rule out a diagnosis of malignancy.
Patient Evaluation
Routine staging investigations include chest x-ray, CT scanning of the abdomen and pelvis, CT scanning of the thorax, and tumor markers. CT scanning of the thorax does not add value in patients with seminoma with no evidence of retroperitoneal lymph node involvement. There is no role for bipedal lymphography in staging patients; however, magnetic resonance imaging (MRI) with lymphotrophic nanoparticles has shown promise in a number of studies but needs further evaluation before it can be adopted into clinical practice.44 In stage II and III seminomas, especially in patients with bulky retroperitoneal disease, a bone scan should also be performed. Abnormal serum marker levels should be monitored to document postorchiectomy decay according to their respective half-lives. Patients with extensive metastatic disease, nonpulmonary visceral metastases (NPVMs), or very high tumor marker levels are at risk for brain metastases, and CT or MRI of the brain should be performed.45 Baseline pulmonary and renal functions are assessed in patients who require chemotherapy.
Tumor Markers
Measurements of AFP, β-HCG, and LDH are essential in the diagnosis and management of patients with GCTs.46 HCG is a glycoprotein with a molecular weight of 45,000 daltons, composed of two subunits, of which the α-subunit is identical to that of luteinizing hormone (LH), follicle-stimulating hormone (FSH), and thyroid-stimulating hormone (TSH), and a distinct β-subunit. HCG is normally produced by the placenta. About 15% of patients with seminoma have elevated β-HCG levels. Low levels of β-HCG may be found in other neoplasms, including prostate, bladder, and renal tumors. The use of marijuana derivatives also may lead to elevated levels. Some cross-reactivity with LH does occur, and in cases where an elevated HCG level is thought to be due to this cross-reactivity, consideration should be given to having levels remeasured after treatment with testosterone.47 The half-life of β-HCG in blood is approximately 22 hours. In the subset of GCT patients with very high levels of HCG, a plateau of this tumor marker is often reached after the fourth cycle of chemotherapy that is considerably greater than normal. Such patients often remain in remission with slowly falling levels of HCG, possibly resulting from tissue binding of this tumor marker. Such persistent elevation of HCG levels following chemotherapy is not in itself an indication for salvage therapy.48
One or both of these markers are elevated in 85% of patients with nonseminomatous GCTs. Experience with surveillance of patients with early-stage disease has shown that normal markers do not exclude the presence of occult disease.49
LDH is another important marker in patients with GCTs. It is elevated in up to 60% of patients with nonseminomas and also in a high proportion of patients with advanced seminomas.42,50
PLAP is an isoenzyme of alkaline phosphatase and is normally expressed by placental syncytiotrophoblasts. It is also expressed by testicular tissue and has been investigated as a tumor marker in seminoma. Although PLAP levels are often elevated in patients with seminoma, this factor has proven to be of little value in clinical management.51
Staging
The 2009 American Joint Committee on Cancer/International Union Against Cancer (AJCC/UICC) TNM classification (Table 53-3) is the recommended staging system. In stage I disease, one of the most important determinants of outcome is the presence of vascular invasion in the primary tumor, and this differentiates a pT1 tumor from a pT2 tumor.52 Tumors with invasion of the spermatic cord are staged as pT3 cancers, and the rare tumors with scrotal invasion are classified as pT4 lesions.

The International Germ Cell Cancer Collaborative Group (IGCCCG) analyzed data from 5168 patients with advanced disease.42 Independent prognostic variables identified by the IGCCCG were included: histologic type (nonseminoma vs. seminoma), site of the primary tumor (testis, retroperitoneal site, or other), presence or absence of NPVMs (brain, bone, or liver), and degree of marker elevation (AFP, β-HCG, and LDH). Based on the results of multivariate analysis, the IGCCCG recommended three prognostic groups for nonseminomatous tumors and two prognostic groups for seminomatous tumors. For nonseminomatous cancers, the 5-year overall survival (OS) for the good-prognosis group was 92%, for the intermediate-prognosis group, 80%, and for the poor-prognosis group, 48%. For seminoma, only two prognostic groups were identified: a good-prognosis group without NPVMs with a 5-year OS of 86% and an intermediate-prognosis group with NPVMs with a 5-year OS of 72%. This classification system has been validated by van Dijk and associates50 using Cox regression analysis and recursive partitioning in a cohort of 3048 NSGCT patients.
Primary Therapy
Surgery
Surgery involves a radical inguinal orchiectomy to allow high division of the spermatic cord. Orchiectomy is both diagnostic and therapeutic by providing adequate tissue to ascertain the diagnosis and offering cure in a high proportion (60% to 90%) of patients with stage I disease.19,53 Although not recommended, an accidental scrotal approach does not appear to compromise the outcome, and no additional therapy is required following scrotal violation, provided that the scrotal cavity has not been grossly contaminated by tumor. In patients with life-threatening metastatic disease and a clear-cut diagnosis of germ cell malignancy, initial management should be with chemotherapy and surgery should be postponed until completion of systemic treatment.19,53 The role of partial orchiectomy in patients with metachronous or synchronous bilateral tumors will be discussed later.
Seminoma
In stage I seminoma, postorchiectomy management options include surveillance, RT, or adjuvant chemotherapy. In the past, the standard management of patients with stage I seminoma has been adjuvant retroperitoneal RT. Although RT provided excellent long-term results, with local control in the abdomen and pelvis in virtually all patients, it is now known that over 85% of patients are cured with orchiectomy alone and that RT has been associated with an increased risk of late gonadal toxicity, development of secondary malignant tumors, and, in some cases, an increased risk of cardiovascular disease.54–58 Various approaches have been tried to reduce radiation morbidity, including a reduced radiation dose and treatment volume. In addition, such alternative management strategies as surveillance (including risk-adapted surveillance) and adjuvant chemotherapy have been investigated.59–63 Although adjuvant retroperitoneal RT constituted the standard of care for the past 50 to 60 years, surveillance is now the standard approach, minimizing the burden of treatment while maintaining the cure rate at 100%.64,65,66
In stage III disease, chemotherapy is the treatment of choice.19,53
Management of Stage I Seminoma
Surveillance
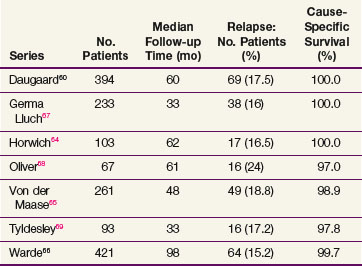
The data on seminoma surveillance are now mature, and relapse rates of 15% to 24% have been reported* (Table 53-4). The two largest reported series are from Copenhagen and Toronto.60,66 In the Copenhagen series of 394 patients, with a median follow-up of 60 months, the crude relapse rate was 17.5%. In the Princess Margaret Hospital, Toronto, series of 421 patients, with a median follow-up of 8.1 years, the actuarial 5-year relapse-free survival (RFS) was 85.5%. The other studies with adequate follow-up (>36 months) have reported similar relapse rates. The predominant site of relapse in all studies was the para-aortic lymph nodes (82% in the Danish Testicular Cancer Study Group (DATECA) study and 89% in the Princess Margaret Hospital series.60,66 The median time to relapse ranged from 12 to 18 months, but occasional late relapses (>4 years) have also been reported.70
Prognostic factors for relapse have been studied in a number of the surveillance studies, and age, tumor size, and small vessel invasion have been reported as predictive of relapse. To more accurately determine prognostic factors for relapse in patients with stage I testicular seminoma managed by surveillance, a pooled analysis of four large surveillance series was performed using individual patient data.71 In 638 patients, tumor size and rete testis invasion predicted for relapse on multivariable analysis. The effect of tumor size on the relapse rate is shown in Figure 53-2, and the hazard ratio for relapse with a tumor size of more than 4 cm was 2 (95% CI, 1.3 to 3.2) relative to baseline (tumor size <4 cm and no rete testis invasion). This model has not been validated in an independent dataset and does not have sufficient discrimination to be clinically useful, because patients in the high-risk group have only a 35% risk of relapse during surveillance. For these reasons, these prognostic factors should not dictate treatment strategy.
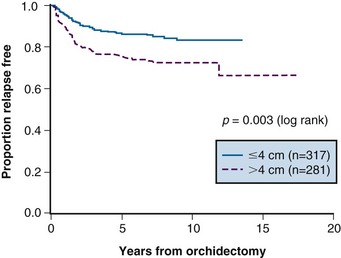
Figure 53-2 Relapse-free rate based on primary tumor size.
From Warde P, Specht L, Horwich A, et al: Prognostic factors for relapse in stage I seminoma managed by surveillance. A pooled analysis. J Clin Oncol 20:4448-4452, 2002. Copyright © American Society of Clinical Oncology.
At relapse, most patients have been treated with retroperitoneal RT. The incidence of second relapse after irradiation is approximately 10%. The likelihood of detecting early progression in the retroperitoneal lymph nodes (nodes <5 cm) and, therefore, suitability of the patient for RT depends on the frequency of follow-up CT scans of the abdomen and pelvis. The current Princess Margaret Hospital follow-up policy is shown in Table 53-5. One of the concerns regarding the routine use of surveillance was the potential for an increased need for chemotherapy. In the Toronto experience, a similar percentage of patients managed with surveillance (4.6%) and adjuvant RT (3.9%) required chemotherapy as part of their management. Therefore, surveillance does not lead to an increased use of chemotherapy, with its ensuing toxicity.
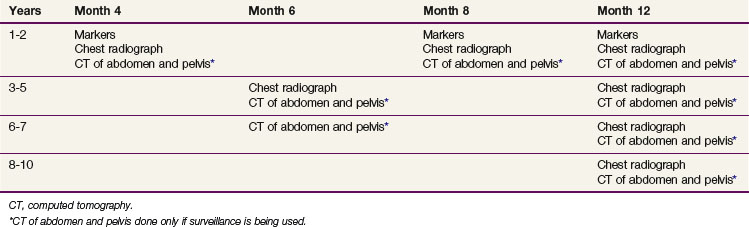
Another concern regarding surveillance relates to the increased use of imaging studies. There are no prospective data available that quantitate the risk of radiation-induced cancer from CT scanning. A recent paper used long-term follow-up data from survivors of the Hiroshima and Nagasaki atomic bombings to estimate the risk of low-level exposure approximating that of one or more CT scans. This approach has been criticized for methodologic reasons. Nonetheless, the seminoma surveillance population is likely at some risk of induced cancer, given the young age of many patients and the use of repeated CT scans of the abdomen. It seems reasonable to attempt to decrease the frequency of imaging as much as possible until more data on the risk of such scans are available. In addition, the use of low-dose CT protocols is currently under study, and these may be used in standard surveillance protocols in the future.72
Radiation Therapy
Overall survival (OS) ranges between 92% and 99% at 5 to 10 years, with the cause-specific survival (CSS) approaching 100%. Most deaths are due to intercurrent illness, but concern exists that premature deaths may be occurring from radiation-induced cancers or cardiac disease.54,58 With modern-era RT, the relapse rate has varied from 0.5% to 5%66,73–77 (Table 53-6). In-field relapse is rare, and when it is suspected, biopsy should be performed to rule out a nonseminomatous tumor or another malignant tumor. The commonest sites of relapse following adjuvant RT are the mediastinum, lungs, and left supraclavicular fossa. A small proportion of patients, usually with predisposing factors, develop relapse in the inguinal nodes. Uncommon sites of isolated metastases such as the brain and tonsils have been noted.45,77 For supradiaphragmatic relapse, chemotherapy is the treatment of choice and results in a cure rate of nearly 100%. Inguinal relapse can often be treated successfully with RT to the involved area.78


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


