(1)
Mayo Clinic, Jacksonville, Florida, USA
4.1 Introduction
Laser photocoagulation was the preferred treatment for the complications of diabetic retinopathy – diabetic macular edema (DME) and proliferative diabetic retinopathy (PDR) – for several decades with level I evidence emerging from the Early Treatment of Diabetic Retinopathy Study [45] and the Diabetic Retinopathy Study (DRS) [166–168]. Laser reduced the risk of severe vision loss (<5/200) in eyes with PDR and moderate vision loss (>15 letters) in eyes with DME by one half, but significant improvements in visual acuity (>15 letters) were unusual.
In an attempt to improve the treatment of macular edema, physicians began injecting triamcinolone acetate into the vitreous of the eyes with macular edema due to diabetic retinopathy and retinal vein occlusions [77, 91, 114] early in the twenty-first century. Corticosteroids improved macular edema dramatically but the high incidences of posterior subcapsular cataracts and elevated intraocular pressures limited their widespread adoption. Surgeons frequently performed pars plana vitrectomy for advanced complications of PDR (vitreous hemorrhage, traction retinal detachment, traction/rhegmatogenous retinal detachment) [169] and even for DME that was accompanied by vitreomacular traction [106]. Surgical outcomes for PDR were superior to the natural history of the disease [169], but vitrectomy only benefited a small number of patients with vision loss due to DME [60].
Effective pharmacotherapy for DME eventually came from a surprising source – tumor biology [24, 56]. The discovery of vascular endothelial growth factor (VEGF) and the early understanding of its role in neovascular age-related macular degeneration, macular edema due to retinal vein occlusions, DME, and PDR [5] set in motion a pharmacologic revolution in ophthalmology. The development of potent ocular pharmaceuticals created a paradigm shift in the treatment of center-involving diabetic macular edema and more recently in the treatment of PDR (Fig. 4.1).
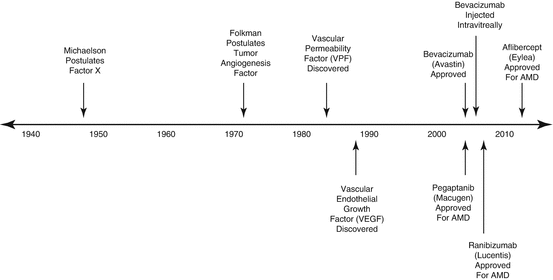
Fig. 4.1
Important milestones in the discovery of vascular endothelial growth factor and the development of VEGF-inhibitory drugs. Each of the VEGF-inhibitory drugs was initially approved by the US Food and Drug Administration for the treatment of neovascular age-related macular degeneration (nAMD) with approvals for diabetic macular edema lagging by 6 years (ranibizumab) and 3 years (aflibercept)
Many manuscripts describing the treatment of DME with anti-VEGF drugs have been published, but most are flawed with small numbers of patients, short durations of treatment and follow-up, lack of control arms and randomization, and inadequate masking. Fortunately, several high-quality, randomized, controlled, double-masked, multicenter trials have been published, and these constitute the focus of this chapter.
4.2 Vascular Endothelial Growth Factor
Pathologic ocular neovascularization has long been recognized as the sequela to severe vascular injury. Michaelson (1948) postulated the existence of a soluble factor X that he believed was responsible for new blood vessel growth in conditions such as neovascular glaucoma (Fig. 4.2) [119]. Folkman (1971) noted that both primary and metastatic solid tumors in children were accompanied by leashes of blood vessels through which the tumor obtained oxygen and nutrients [61]. He postulated that solid tumors synthesized a pro-angiogenic substance that promoted blood vessel growth necessary to sustain tumors.
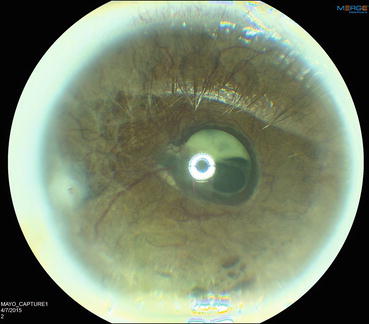
Fig. 4.2
Slit lamp photograph showing severe neovascularization of the iris. Contraction of the neovascular membrane on the anterior surface of the iris everts the pupillary margin and exposes the posterior pigmented epithelium of the iris (ectropion uvea)
Vascular permeability factor (VPF) was discovered in 1983 [155], and Ferrara and Connolly independently discovered vascular endothelial growth factor (VEGF) [25, 56] 6 years later. Sequencing analysis showed that both investigators had identified the same molecule, which turned out to be the previously discovered VPF. This set in motion a multi-specialty, international research initiative that characterized VEGF, its receptors, and its downstream pathways.
VEGF is actually a group of closely related molecules that segregate into seven families: VEGF-A, VEGF-B, VEGF-C, VEGF-D, the orf virus encoded VEGF-E, VEGF-F, and placental growth factor (PlGF) (Table 4.1). Most of these families consist of several isoforms with similar binding sequences.
Table 4.1
The various VEGF families, the most important isoforms, and their major physiologic effects
Vascular endothelial growth factors and their major functions | |
|---|---|
VEGF-A (VEGF121, VEGF145, VEGF165) | Responsible for most angiogenesis in adults Responsible for most ocular neovascularization and breakdown of the blood retinal barrier |
VEGF-B | Responsible for coronary angiogenesis Promotes tumor metastasis Found in choroidal neovascular membranes |
VEGF-C | Main function is lymphangiogenesis Found to participate in angiogenesis, perhaps including choroidal neovascular membranes Participates in neural development and blood pressure regulation |
VEGF-D | Main function is lymphangiogenesis surrounding pulmonary bronchioles |
VEGF-E | Encoded by orf viruses |
VEGF-F | Expressed in the venom of the habu snake |
Placental growth factor 1 and 2 | Found in choroidal neovascular membranes Important for vasculogenesis and angiogenesis during ischemia, inflammation, wound healing, and cancer |
VEGF-A isoforms are the most important contributors to ocular angiogenesis. The 14 kb VEGF-A gene resides on chromosome 6p21.3 [174]. Splicing of eight exons and seven introns creates at least six major (VEGF121, VEGF145, VEGF165, VEGF183, VEGF189, and VEGF206) and several minor isoforms within the VEGF-A family [84, 170]. In addition, the split product VEGF110 is produced when tissue-sequestered isoforms with 165 or more amino acids are cleaved by plasmin and matrix metalloproteinase-3. The shorter non-heparin-binding isoforms are biologically active but with only 10–20% the biological activity of VEGF165. VEGF naturally exists as a homodimer, so it presents two receptor-binding sites to its environment [95, 104].
Each of the VEGF families controls a different type of angiogenesis (Table 4.1). VEGF-B stimulates coronary blood vessel growth, and VEGF-C and VEGF-D promote lymphangiogenesis by activating VEGF receptor (VEGFR) 3 [94]. The exact role of PlGF in angiogenesis is unclear, but its binding to VEGFR1 may force VEGF-A to preferentially bind to VEGFR2 [137], the receptor primarily responsible for ocular angiogenesis. PlGF may play an active role in the development of DR as it has been found in the retinas of diabetic rats [89, 99].
The common receptor-binding sequence of all VEGF-A isoforms resides in the amino acid region 82–93, whereas the heparin-binding sequence is found in the 111–165 amino acid region of longer isoforms. Molecular charge determines isoform diffusibility with the acidic VEGF121 isoform being freely diffusible whereas the basic VEGF206 isoform is nearly totally bound to interstitial matrix. Fifty to seventy percent of the electrically neutral VEGF165 is bound to the interstitial matrix, and heparin-binding sequences enable isoforms of 165 amino acids and longer to bind to interstitial matrix heparin moieties. Sequestration of these longer molecules provides a readily available reservoir of VEGF when injury or inflammation induces a protease-mediated breakdown of the interstitium. The heparin-binding domain also enables VEGF165 to bind to the transmembrane neuropilin-1 co-receptor.
Diffusible VEGF binds to the extracellular binding domains of three transmembrane receptors: VEGFR1 (flt-1), VEGFR2 (flk-1), and VEGFR3 (flt-4). Each of these receptors is composed of three major parts: the seven extracellular immunoglobulin-like VEGF-binding domains, the single transmembrane region, and the intracellular tyrosine kinase moieties [28, 165]. VEGFR1 binds VEGF-A, VEGF-B, and placental growth factor; VEGFR2 binds VEGF-A and VEGF-C; and VEGFR3 binds VEGF-C, VEGF-D, VEGF-E, and VEGF-F. VEGFR-1 binds VEGF-A with a much higher affinity than does VEGFR2. Binding of VEGF by VEGFR1 and VEGFR2 occurs at the second and third extracellular domains. Alternative splicing of the RNA transcript for the binding domains of the VEGF receptors creates soluble VEGFR2 (sVEGFR2) and VEGFR1 (sVEGFR1), which inhibit vascular development by promoting vascular maturation and maintaining corneal avascularity [137].
The role of VEGFR1 in angiogenesis is unclear because its downstream activity varies with both the age of the individual and the cell type. Activation of VEGFR1 during embryological development stimulates angiogenesis, but during adulthood it dampens VEGF activity by acting as a decoy receptor.
VEGF-A activity occurs when either naturally occurring VEGF121 or VEGF165, or the fibrin-split product VEGF110, binds to VEGFR2 (Fig. 4.3). Each of two VEGFR2 receptors binds to the VEGF dimer causing conformational changes in the 3-dimensional structures of the receptor molecules and activating their intracytoplasmic tyrosine kinase moieties (Fig. 4.2). This phosphorylates several intracellular enzymes that activate downstream pathways, which lead to VEGF’s potent effects (Fig. 4.3) [21, 48, 79, 164].
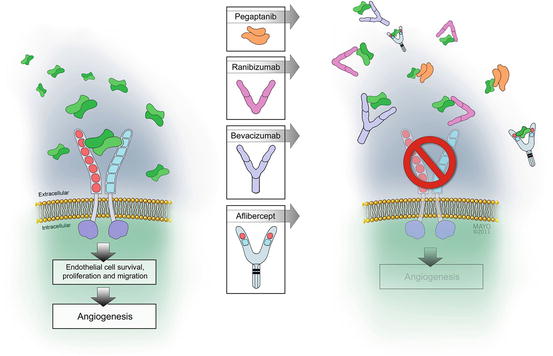
Fig. 4.3
Diffusible dimers of vascular endothelial growth factor (VEGF) bind to transmembrane receptors causing them to dimerize. Receptor dimerization leads to conformational changes and activation of the cytoplasmic tyrosine kinase moieties. These phosphorylate several cytoplasmic proteins, which upregulate downstream biochemical pathways. Each of the four available VEGF-inhibitory drugs binds to diffusible VEGF to prevent receptor binding
Hypoxic environments created by processes such as tumor growth and occlusive vasculopathies upregulate VEGF [42]. Tissue hypoxia reduces hydroxylation of the cell’s “oxygen sensor” HIF-1α, preventing it from binding to the von-Hippel Lindau factor, and undergoing ubiquination and subsequent destruction within proteosomes. Stable HIF-1α dimerizes with HIF-1β to form a complex that enters the cell nucleus where it activates the promoter region of the VEGF gene and also upregulates both VEGFR1 and VEGFR2 [71, 172] synthesis (Fig. 4.4).
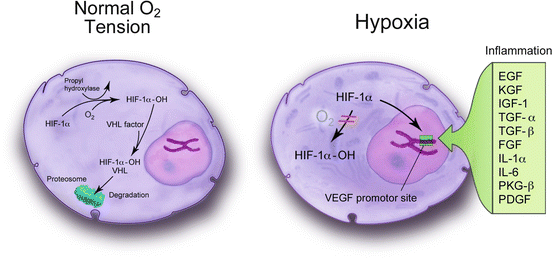
Fig. 4.4
Hypoxia inducible factor (HIF)-1α is normal tissue oxygen conditions and is subsequently metabolized in the proteosomes. Under hypoxic conditions and in the presence of various chemokines and cytokines, HIF-1α becomes stabilized by dimerizing with HIF-1β, enters the cell nucleus, and binds to the promoter region of the vascular endothelial growth factor gene
VEGF is upregulated during starvation and by several metabolic factors, chemokines, and cytokines. A variety of metabolic regulators including ROS (reactive oxygen species such as superoxide (O2 −)) increase the synthesis of both VEGF and its receptors [173]. Several growth factors including epidermal growth factor, tumor necrosis factor (TNF)-α, transforming growth factor (TGF)-β, basic fibroblast growth factor, interleukin-6, insulin-like growth factor-1, keratinocyte growth factor, and platelet-derived growth factor upregulate VEGF and may work in conjunction with tissue hypoxia [128]. Oncologic mutations that stimulate RAS, a family of ubiquitous intracellular GTP-ases, upregulate VEGF [78]. Mechanical forces such as sheer stress and stretch that occur with vitreomacular traction promote VEGF production [46].
VEGF has been detected in both the neuroretina and pigment epithelium [97], and it can be produced by several cell types including pericytes, glia, vascular endothelial cells, invading leukocytes, and retinal pigment epithelium [2, 134]. Some VEGF isoforms modulate the synthesis of others by affecting HIF-1α stability [183], and VEGF inhibitors probably function via a similar mechanism.
PlGF and VEGF-B binding to VEGFR1 activates signaling pathways implicated in monocyte chemotaxis [36] and inflammatory-related angiogenesis. Though PlGF appears to be a critical mediator of inflammatory angiogenesis, it does not appear to directly influence embryologic or physiologic adult angiogenesis [59]. Tyrosine kinase activity of VEGFR1 is relatively weak but several downstream signaling molecules including phospholipase C, the p85 subunit of PI-3 kinase, growth factor receptor bound protein, SHP-2, and NcK are associated with VEGFR1 phosphorylation sites. These pathways result in eNOS activation and calcium influx in vascular endothelial cells.
VEGFR2 activation is responsible for the full spectrum of VEGF-A-mediated responses within human endothelial cells (survival, proliferation, migration, and formation of a vascular tube) by stimulating Raf-MEP-ERK and MAPK pathways [164]. These pathways phosphorylate several endothelial cell proteins including phospholipase-3, PI-3 kinase, RAS GTP-ase activating protein and the Src family [48, 79]. They stimulate cell migration via FAK and paxillin, PI-3 kinase, and MAPK and prolong survival via PI-3 kinase and Akt/PKB [21].
VEGF-A isoforms also bind to the heparin-binding domains of neuropilin-1 and neuropilin-2, transmembrane proteins with small, non-catalytic tails that present VEGF165 favorably to VEGFR2. The neuropilin-modulated binding of VEGF165 increases its biological activity by tenfold over that of VEGF121, which cannot interact with neuropilin [63].
VEGF-A upregulates gene expression that promotes the division, migration, and survival of vascular endothelial cells, regulates vascular dilation and permeability, and stabilizes immature blood vessels (Fig. 4.5) [55]. VEGF-A enhances vascular permeability 50,000 times more than does histamine, increases nitric oxide production, and stimulates the synthesis of several molecules that are important to angiogenesis, such as matrix metalloproteinases, which break down the interstitial matrix to allow advancement of proliferating endothelial cells [175]. VEGF165 induces endothelial cell growth by activating the Raf-MEK-Erk pathway [52]. VEGF degrades the blood retinal barrier (BRB) in venules but not capillaries [126] by causing a calcium-mediated increase in hydraulic conductivity [14], increase in endothelial cell fenestrations [147], and opening of intercellular tight junctions by phosphorylating several junctional proteins [67]. Increased permeability allows plasma to extravasate and form an interstitial fibrin gel into which endothelial cells can proliferate and migrate [44]. VEGF-A increases hexose transport to meet the metabolic demands of neovascularization [138]. VEGF-A produces a dose-dependent vasodilation in vitro via nitric oxide synthesis [102] and retinal vasodilation in animal models and human retinal vein occlusions [51, 171].
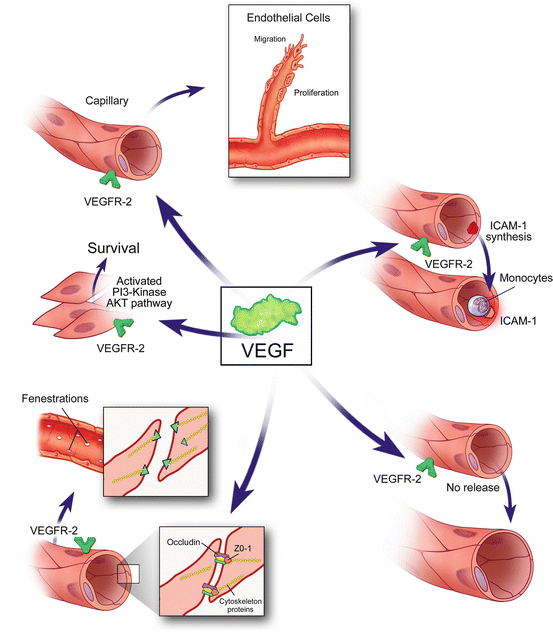
Fig. 4.5
The many functions of vascular endothelial growth factor A
VEGF-A promotes the survival of rat endothelial cells in vitro and in vivo, while VEGF blockade causes extensive apoptotic changes in the vasculature of neonatal but not adult mice [73]. Newly formed but not mature vessels are VEGF dependent [16, 181], and a complete vestment of pericytes is key to establishing VEGF independence [16]. Nonetheless, VEGF-A plays a role in maintaining the choriocapillaris in adults [53]. VEGF-A administration to hypoxic vascular endothelial cells upregulates phosphatidylinositol (PI)-3 kinase-Akt pathways and the expression of antiapoptotic proteins Bl-2 and A1 and prevents apoptosis and regression of the vasculature [6, 72]. The effects of VEGF on the retinal pigment epithelium remain controversial as some investigators have noted breakdown of the outer blood-retinal barrier whereas others have not [1, 74, 85, 122].
VEGF-A promotes monocyte chemotaxis and colony formation by granulocyte-macrophage progenitor cells. VEGF-A upregulates the synthesis of intercellular adhesion molecule (ICAM)-1 and vascular cellular adhesion molecule (VCAM)-1, which allows natural killer (NT) cells to adhere to vascular endothelial cells.
Exogenous administration of VEGF into primate eyes induces changes that resemble diabetic retinopathy [171]. VEGF has been successfully blocked in animals with soluble decoy receptors [93], tyrosine kinase inhibitors, and antibodies directed against the VEGF dimer or its receptors [101]. In animal models, blocking VEGF pathways appears to be more effective than blocking other angiogenic pathways [52]. Drugs that block VEGF work primarily by reestablishing the blood-retinal barrier, “normalizing” the circulation, and preventing further new blood vessel growth.
4.3 Anti-VEGF Drugs
Currently available drugs that inhibit the actions of vascular endothelial growth factor are listed in Table 4.2. Included are the major binding and pharmacokinetic properties of each drug.
Table 4.2
Drugs that bind to and inhibit the actions of vascular endothelial growth factor that have been used to treat ocular angiogenesis
Important characteristics of drugs that bind vascular endothelial growth factor | ||||
|---|---|---|---|---|
Characteristic | Aflibercept | Bevacizumab | Pegaptanib | Ranibizumab |
Description | Fusion protein with receptor sequences bound to Fc fragment of IgG | Recombinant, humanized, murine antibody to VEGF-A | Pegylated aptamer | Recombinant, humanized, murine antibody fragment to VEGF-A |
Molecular weight (kDa) | 115 | 149 | 50 | 48 |
Isoforms and families bound | VEGF-A, VEGF-B, placental growth factor | VEGF-A | VEGF165 | VEGF-A |
Binding affinity for VEGF165 (pmol) | 0.5 | 58–1000 | 46–192 | 50 |
Intravitreal half-life (days) | 9 (estimated) | 9.8 | 7 (estimated) | 7.1 |
Serum half-life (days) | 6 | 21 | 0.2 | 2–4 |
4.3.1 Pegaptanib
Pegaptanib is a 28-base oligonucleotide – a modified RNA fragment or aptamer – that attaches to the heparin-binding domain of VEGF165. It has a molecular weight of 50 kDa, a negative charge of 28, and a hairpin structure. The addition of a 40 kDa polyethylene glycol chain (pegylation) increased the drug’s binding affinity (K D = 50 pM for VEGF165) and stability by protecting it against the action of endonucleases [152]. After injection into monkey eyes, the intravitreal concentration of pegaptanib decreases according to a first-order kinetics model with an intraocular half-life of 94 h. Pegaptanib accumulation in the serum is limited by both metabolism (endo- and exonuclease actions) and urinary excretion.
Drug developers had been concerned that pan-VEGF suppression might increase the risk of serious systemic adverse events so they developed pegaptanib to inhibit the actions of diffusible VEGF165 but not the shorter natural VEGF121 and fibrin split product VEGF110. Pegaptanib’s exact mechanism of action is not fully known, but it may prevent VEGF binding to neuropilin-1 (by obstructing the heparin-binding site), or it may stereoscopically interfere with VEGF’s receptor-binding site. Pegaptanib reduces endothelial cell proliferation [15], completely inhibits VEGF-mediated vascular permeability in a guinea pig model, and reduces experimental neovascularization, leukostasis, and BRB breakdown in diabetic rats [86]. In vitro studies suggest that pegaptanib inhibits VEGF-mediated processes as effectively as monoclonal antibodies [50]. A preclinical study in rhesus monkeys determined that several doses of pegaptanib were safe, and intraocular samples obtained 7 and 28 days after intravitreal injections demonstrated no molecular instability [43].
The important pegaptanib drug trials and their key findings are listed in Table 4.3.
Table 4.3
Important trials with pegaptanib and details their key findings
Important diabetic macular edema trials with pegaptanib | ||
|---|---|---|
Trial and phase | Cohorts | Key findings |
Macugen DME trial Phase II 172 patients | Pegaptanib 0.3 mg 1.0 mg 3.0 mg Sham | At 36 weeks, patients treated with 0.3 mg pegaptanib compared to sham/laser: 1. Greater likelihood of 10-letter gain (34% vs. 10%, P = 0.003) 2. Trend toward 15-letter gain (18% vs. 7%, P = 0.12) 3. Greater improvement in mean BCVA (+4.7 vs. −0.4 letters, P = 0.02) 4. Greater improvement in macular thickness (−68 μm vs. +4 μm, P = 0.02) |
Querques et al. Retrospective 63 patients | Single cohort | At 36 weeks compared to baseline, patients had significant improvements in: 1. BCVA (P < 0.019) 2. Macular thickness (P < 0.001) |
Macugen study group Phase II/III 260 patients | At 102 weeks, patients treated with 0.3 mg pegaptanib compared to laser: 1. Greater likelihood of 10-letter gain (36.8% vs. 19.7%, P = 0.0047) 2. Trend toward 15-letter gain (16.5% vs. 10.2%, P = 0.2466) 3. Greater improvement in mean BCVA (+6.1 vs. +1.3 letters, P < 0.01) 4. Fewer patients required laser for persistent edema (25.2% vs. 45.5%, P = 0.003) | |
The VEGF Inhibition Study in Ocular Neovascularization Clinical Trial Group (VISION) trials led to United States Food and Drug Administration (US FDA) approval of pegaptanib for the treatment of neovascular age-related macular degeneration (AMD). Compared to observation, patients receiving injections of 0.3 mg pegaptanib every 6 weeks experienced only half as much vision loss at 1 year (−14 vs. −7 letters) [76]. In the post-approval Evaluation of Efficacy and Safety in Maintaining Visual Acuity with Sequential Treatment of Neovascular AMD (LEVEL) trial, maintenance therapy with pegaptanib sustained most of the gains in vision previously acquired with the more potent, nonselective VEGF inhibitors bevacizumab and ranibizumab [62].
Following the nAMD trials, pegaptanib underwent clinical testing for the treatment of DME. In a phase II trial, 172 patients were randomized to receive one of three pegaptanib doses (0.3 mg, 1 mg, or 3 mg) or sham injections every 6 weeks for 36 weeks. A greater proportion of patients receiving 0.3 mg pegaptanib than sham improved by at least 10 letters (34% vs. 10%; P = 0.003) and at least 15 letters (18% vs. 7%; P = 0.12). Patients receiving 0.3 mg pegaptanib as compared to sham experienced a greater mean improvement in BCVA (+4.7 vs. −0.4 letters; P = 0.04) and greater mean decrease in macular thickness (−68 μm vs. +4 μm; P = 0.02). One case of endophthalmitis occurred after 652 injections [113].
In a 6-month retrospective study of 63 eyes with DME, patients receiving pegaptanib experienced significant improvements in BCVA (P = 0.019) and macular thickness (P < 0.001) compared to baseline. Patients were treated every 6 weeks PRN for recurrent edema, and most eyes required at least three injections. Compared to the phase II trial, patients in this retrospective study experienced similar rates of 1-line improvement in vision (55.6% vs. 59%) and reduction in CRT (42% vs. 29.6%) [143].
A phase II/III trial randomized 260 patients to receive pegaptanib or sham injections every 6 weeks [163]. Significantly more patients receiving pegaptanib than sham injections gained at least 10 letters (36.8% vs. 19.7%; P = 0.0047) though the proportions gaining at least 15 letters were not significantly different (16.5% vs. 10.2%; P = 0.2466). At week 102, patients receiving pegaptanib had greater mean gains in VA (+6.1 vs. +1.3 letters; P < 0.01), and fewer pegaptanib-treated patients required grid laser photocoagulation by week 54 (23.3% vs. 41.7%; P = 0.002) and week 102 (25.2% vs. 45.0%; P = 0.003) for persistent edema. A slightly higher proportion of pegaptanib-treated patients experienced a 25% reduction in CRT. Pegaptanib appeared safe since only two patients suffered cerebrovascular accidents, compared to one patient in the sham group.
Self-reported quality of life (QoL) was assessed with the 25-item National Eye Institute Visual Function Questionnaire (NEI VFQ 25) and the EQ-5D [109]. The NEI VFQ 25 domains of near vision, distance vision, and social functioning (week 54) and distance vision, social functioning, mental health, and composite score (week 102) demonstrated clinically meaningful (>5-point between-group difference) and statistically significant (P < 0.05) benefits favoring pegaptanib. No significant difference in the mean change in generic EQ-5D-weighted utility scores was seen. The authors concluded that the VA improvement from pegaptanib treatment versus sham is accompanied by improved vision-related QoL as reported by the DME patient.
Pegaptanib causes marked regression of new vessels in eyes with proliferative diabetic retinopathy (PDR) [3, 13, 101, 116]. Pegaptanib was evaluated as an adjunct to vitrectomy in eyes with recurrent vitreous hemorrhage due to PDR. Fifteen eyes of 14 patients each received one to three pegaptanib injections prior to undergoing vitrectomy for persistent vitreous hemorrhage (VH) or progressive traction retinal detachment. In the majority of patients with VH, pegaptanib enabled surgeons to place additional laser prior to vitrectomy. Surgery after pegaptanib injections was felt to be faster and less challenging compared with conventional vitrectomy for recurrent VH due to PDR [83].
Pegaptanib showed promise in the early DME trials but further development was halted because it was inferior to the pan-VEGF-A blockers bevacizumab and ranibizumab for the treatment of nAMD. The drug’s developer appeared unwilling to devote the resources necessary to run large, multicenter, phase III DME trials. Some authors have argued that VEGF165-specific binding may have doomed pegaptanib for the treatment of nAMD because it was unable to prevent the pro-angiogenic effects of VEGF121 and VEGF110. Since these isoforms appear to be less important in the development of DME, pegaptanib may have been relatively more effective in this condition. The theoretical safety advantages of VEGF165 specific binding over pan-VEGF-A binding in a diabetic population that is at risk of Antiplatelet Trialists’ Collaborative (APTC)-defined events may also have favored pegaptanib use in patients with DME. Unfortunately, pegaptanib never received US FDA approval for DME, and plans for registration trials are unlikely to be resurrected. Its frequency of use in patients with nAMD and DME remains insignificant.
4.3.2 Bevacizumab
Bevacizumab is a humanized (93% of protein sequences are human), recombinant, murine-derived, monoclonal antibody that binds all isoforms of VEGF-A. Bevacizumab has an impressive dissociation constant for VEGF165 (K D = 56–1100 pM) though its binding affinity is less than that of the approved anti-VEGF drugs [136, 140]. Bevacizumab maximally inhibits proliferation of human umbilical vascular cells and improves their survival when the bevacizumab/VEGF ratio exceeds 2.6:1 [176].
The intravitreal half-life of bevacizumab in human eyes varies between 6.7 and 9.82 days, with an average of 8.25 days [117, 161]. Very poor intraocular penetration occurs after topical administration as only small quantities of drug reach the iris, ciliary body, and retina. Subconjunctival and intravitreal injections in rabbits produce similar serum concentrations, suggesting that the drug passes into the systemic circulation without being altered within the eye. Bevacizumab possesses a long serum half-life (21 days) because of its Fc fragment, and it significantly lowers serum VEGF concentrations [10].
Bevacizumab reaches the subretinal space in rabbit eyes within 2 h after intravitreal injections and reaches the inner retina, choroid, and serum in monkeys within the first day [35, 81, 156]. The peak concentrations in the iris and retina are twice those in the vitreous, suggesting that the drug undergoes active transport and binding within the tissues. Bevacizumab can be internalized by RPE cells though toxicity has not been noted [81].
Bevacizumab was developed for the systemic treatment of solid tumors [57] and is currently approved for advanced colorectal carcinoma, small cell lung cancer, renal cell carcinoma, cervical carcinoma, ovarian carcinoma, and glioblastoma. In a small pilot study, bevacizumab was administered intravenously to patients with nAMD [120]. Patients experienced anatomic and functional improvements, but the incidence of systemic side effects precluded further use. In 2005, bevacizumab was injected intravitreally into patients with macular edema due to central retinal vein occlusion and nAMD [150, 151].
The important bevacizumab diabetic retinopathy trials and their key findings are listed in Table 4.4.
Table 4.4
The important diabetic retinopathy trials with bevacizumab and their key findings
Important diabetic macular edema trials with bevacizumab | ||
|---|---|---|
Trial and phase | Cohorts | Key findings |
DRCR.net Phase II | Bevacizumab 1.25 mg 2.5 mg Laser | Q6wk injections, 12-week endpoint Bevacizumab-treated patients averaged one-line BCVA gain better than those receiving laser Bevacizumab patients had 11% improvement in macular thickness at week 3, but stable through week 12 |
Pan-American Collaborative Retina Group 139 eyes | Bevacizumab 1.25 mg 2.5 mg | 24 month, retrospective study Patients receiving 1.25 mg and 2.5 mg had improved 1. BCVA (20/150 to 20/75, P < 0.0001; 20/168 to 20/114, P = 0.02) 2. Central macular thickness (466.5 μm to 286.6 μm, P < 0.0001; 423.4 μm to 271.8 μm, P = 0.001) |
Soheilian et al. (2009) 150 eyes Prospective | 3 treatment arms: Bevacizumab Bevacizumab + IVT Laser | At 36 weeks 1. 2 lines improvement in BCVA in 37, 25, and 14.8% of eyes. 2. Significant improvements in central retinal thickness only at 6 weeks |
BOLT study Phase II 80 eyes Prospective | 2 treatment arms: Bevacizumab q6wk Laser q4month | At 1 year 1. More eyes receiving bevacizumab improved by 15 letters (11.9% vs. 5.3%) 2. More eyes receiving bevacizumab improved by ten letters (31% vs. 7.9%) 3. Bevacizumab eyes had greater improvements in mean BCVA (+8.0 vs. −0.5 letters, P = 0.0002) At 2 years 1. More eyes receiving bevacizumab improved by 15 letters (32% vs. 4%) 2. More eyes receiving bevacizumab improved by ten letters (49% vs. 7%) 3. Bevacizumab eyes had greater improvements in mean BCVA (+8.6 vs. −0.5 letters) |
The DRCR.net evaluated the short-term (12-week primary endpoint) efficacy of q6week bevacizumab in a phase II DME trial. One hundred twenty-one patients were randomized to receive laser photocoagulation or intravitreal injections of 1.25 mg or 2.5 mg bevacizumab, with or without laser. Patients receiving bevacizumab experienced a one-line improvement in visual acuity compared to those treated with laser. Approximately one-half of the bevacizumab-treated patients experienced a greater than 11% decrease in macular thickness at week 3, but additional improvements through week 12 were not seen. The authors concluded that 6-week injection intervals may be too long and that combining bevacizumab with laser photocoagulation provides no short-term advantage over bevacizumab monotherapy [30].
The Pan-American Collaborative Retina Study (PACORES) reported the results of a 24-month retrospective study of 139 eyes (115 patients) with DME [8]. Patients who received 1.25 mg or 2.5 mg bevacizumab experienced significant improvements in mean VA (20/150 to 20/75, P < 0.0001; 20/168 to 20/114, P = 0.02) and central macular thickness (466.5 μm to 286.6 μm, P < 0.0001; 423.4 μm to 271.8 μm, P = 0.001).
In a 36-week prospective study, 150 eyes with DME were randomized to receive bevacizumab monotherapy (q12week injections), bevacizumab in combination with intravitreal triamcinolone, or macular laser photocoagulation [158]. Two Snellen lines of visual acuity improvement were achieved by 37% (bevacizumab monotherapy), 25% (bevacizumab + triamcinolone), and 14.8% (laser) of eyes. Compared to baseline, VA improved significantly at all visits in the bevacizumab monotherapy group, only at weeks 6 and 12 in the combination therapy group, but at no visits in the laser group. Significant improvements in central retinal thickness were seen only at 6 weeks. The authors concluded that quarterly bevacizumab injections are superior to laser photocoagulation but that triamcinolone provides no added benefit.
The Bevacizumab Or Laser Therapy (BOLT) in the management of diabetic macular edema trial best demonstrated the superiority of bevacizumab over laser for the treatment of DME [118, 157]. This prospective, single-center, 2-year trial randomized 80 eyes to receive q6week PRN bevacizumab or q4week PRN laser. At 1 year, more patients receiving bevacizumab than laser gained >15 letters (11.9% vs. 5.3%) and >10 letters (31% vs. 7.9%), and fewer lost >15 (2.4% vs. 26.3%) and >30 (0% vs. 5.3%) letters. Patients receiving bevacizumab experienced greater mean VA improvements compared to laser at 1 year (+8.0 vs. −0.5 letters, P = 0.0002) and 2 years (+8.6 vs. −0.5 letters). At 2 years, 49% of bevacizumab-treated patients improved by at least +10 letters and 32% by at least +15 letters, compared to only 7 and 4% of laser-treated eyes. Fewer bevacizumab than laser patients lost 15 letters (0% vs. 14%, P = 0.03). Eyes receiving bevacizumab had greater mean decreases in macular thickness compared to laser (−146 μm vs. −118 μm). The median number of treatments through 2 years was 13 bevacizumab injections and 4 laser treatments.
A post hoc analysis of the BOLT data showed that eyes with subretinal fluid at baseline were most likely to have persistent edema at 2 months [157], and the authors noted that resolution of edema by 4 months is a strong predictor of a favorable long-term response. They found that 20% of eyes with persistent edema at 12 months achieved dry retinas at 24 months with VA improvements of at least 15 letters. They stated that although the 4-month response may be predictive of long-term outcome, it should not lead to withholding of therapy.
Intravitreal injections of bevacizumab cause regression of optic disk neovascularization due to PDR [9, 159], but the effect is transient as neovascularization recurs by 12 weeks after single intravitreal injections [92]. Several studies have reported resolution of chronic vitreous hemorrhage after intravitreal injections of bevacizumab [4, 29, 47, 123], but these results must be viewed with caution since bevacizumab likely does not promote the clearing of vitreous hemorrhage, but rather it allows hemorrhage to clear while it prevents further bleeding.
Intravitreal injections of bevacizumab have been used as adjuvants to improve surgical outcomes by decreasing intraoperative hemorrhage, facilitating fibrovascular membrane dissection (Fig. 4.6) [22, 27, 29, 87, 108, 123, 135, 146, 148, 180] and reducing the incidence of postoperative vitreous hemorrhage [4, 29, 148, 179]. These studies, however, were limited by relatively small numbers of patients, heterogeneous retinal pathology (TRD and VH were studied together), and varying surgical techniques (multiple surgeons and different gauge vitrectomies). Many surgeons remain concerned that the preoperative administration of bevacizumab may worsen fibrovascular traction [7, 90, 124, 135] and may cause enlargement of the foveal vascular zone [23, 104, 105]. Some surgeons recommend that bevacizumab should be administered only within a few days of planned surgery so that prompt action can be taken if a traction retinal detachment worsens or a traction-rhegmatogenous detachment develops.
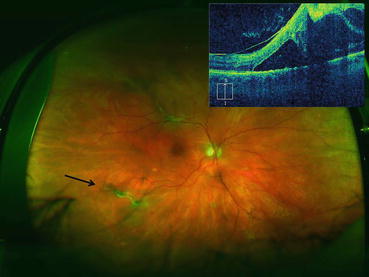
Fig. 4.6
Ultra-widefield photograph showing an eye with proliferative diabetic retinopathy. Fibrovascular proliferation can be seen along the superotemporal arcade causing a traction retinal detachment that threatens the fovea. The vertical optical coherence tomography scan (upper left corner of image) was displaced superiorly to better demonstrate the traction retinal detachment in the superior macula that threatens the fovea. Fibrovascular proliferation just outside the inferotemporal arcade features active neovascularization (black arrow). An intravitreal injection of bevacizumab was given 3 days prior to pars plana vitrectomy to reduce intraoperative bleeding
In a much larger study, 99 eyes of 90 patients scheduled for diabetic vitrectomy were randomized to receive intravitreal bevacizumab preoperatively or no injection [139]. Thirty-four patients received IVB on an average of 11.5 (range, 3–30) days before vitrectomy. Visual acuity improved significantly after surgery in both groups: from 20/617 to 20/62 in the IVB group, and from 20/443 to 20/86 in the non-IVB group (P = 0.11 between groups). Surgery time and postoperative complications (glaucoma, RD, and repeat vitrectomy rate) were similar in both groups. In patients under the age of 40 years, operating time was shorter in the bevacizumab group (P = 0.02) with a trend toward better VA. The authors concluded that preoperative bevacizumab may be a useful adjunct to vitrectomy for severe PDR complicated by TRD, particularly in younger patients.
A meta-analysis of randomized controlled trials compared the safety and functional outcomes of vitrectomy with or without preoperative intravitreal bevacizumab for PDR [182]. Eight trials with 414 eyes of 394 patients were included. The authors found that vitrectomy with preoperative bevacizumab shortened overall surgical time (mean difference of 26.89 min, 95% confidence interval (CI) 31.38–22.39; P < 0.00001) and reduced the number of required endodiathermy applications (mean difference of 3.46, P = 0.02) compared to vitrectomy alone. The bevacizumab group also experienced less intraoperative bleeding (odds ratio [OR] 0.10; 95% CI 0.02–0.46; P = 0.003) and recurrent vitreous hemorrhage within the first month (OR 0.35; 95% CI 0.21–0.58; P < .0001), but the incidence of recurrent vitreous hemorrhage after the first month was comparable between the two groups. There were no significant differences in other complication rates between the two groups, except for iatrogenic retinal breaks, which were more likely to occur in the vitrectomy-alone group (OR 0.27, 95% CI 0.12–0.63; P = 0.003).
4.3.3 Ranibizumab
Scientists at Genentech recognized that VEGF suppression could be used to treat nAMD, DME, and macular edema due to retinal vein occlusions, but they worried that a full-length antibody such as bevacizumab might have limited efficacy and an unfavorable safety profile. Monkey studies showed that the full-length HER2 antibody (molecular weight 149 kDa) was unable to cross the inner retina after intravitreal injection [125], an observation consistent with the proposed inner retina exclusion limit of 76 kDa [88].They also worried that the antibody’s Fc fragment might incite intraocular inflammation or cause systemic adverse events because of a prolonged serum half-life. For these reasons, ranibizumab, an affinity enhanced, humanized, single binding site antibody fragment (Fab), was created in 1996 [54] from a murine antibody to human VEGF. Ranibizumab (MW, 48 kDa) binds all isoforms of VEGF-A, but with a molar affinity for VEGF165 that is 5–20 times that of bevacizumab. Its dissociation constant for the binding of VEGF165 is 44–192 pM [111, 136]. After successfully completing phase III trials [17, 149], ranibizumab was approved by the US FDA in 2006 for the treatment of neovascular AMD.
Ranibizumab concentrations in the retina are 2.2- to 3-fold lower than in the vitreous [70] but since they are 3000-fold greater than VEGF concentrations, maximum binding of VEGF probably occurs within the retina. The terminal intraocular half-life of ranibizumab in monkey eyes appears to be concentration dependent, ranging from 2.63 days (0.5 mg dose) to 3.95 days (2.0 mg dose) [69]. The intravitreal half-life of ranibizumab in rabbits is slightly shorter at 2.1–3 days [70]. In human eyes, the intravitreal half-life ranges from 7.1 days in a study that sampled aqueous concentrations to 9 days in a population kinetics study [96, 178]. Ranibizumab appears to pass unaltered through the trabecular meshwork and choroid into the systemic circulation where it has a half-life of only 2 h because of rapid ultrafiltration by the kidneys. Serum concentrations are 10,000-fold lower than those in the vitreous [68], and ranibizumab does not appear to suppress serum concentrations of VEGF [10].
Ranibizumab reduces the VEGF-mediated in vitro proliferation of human vascular endothelial cells (HUVAC) in a dose-dependent manner [110] and inhibits VEGF-mediated vascular permeability in guinea pigs. Both ranibizumab and bevacizumab increase vascular endothelial cell apoptosis and decrease their proliferation, migration, and assembly into vascular structures [26]. Both drugs decrease VEGF expression, VEGFR2 phosphorylation, and Akt expression [26]. Ranibizumab causes a greater reduction in endothelial cell proliferation whereas bevacizumab has a greater effect on migration, tube formation, and VEGFR2 phosphorylation [26]. In vitro proliferation of pig choroidal endothelial cells is decreased by ranibizumab (44.1%), bevacizumab (38.2%), and pegaptanib (35.1%) [160].
Pharmacokinetic studies suggested that clinically achievable ranibizumab concentrations within the retina after intravitreal injections were capable of suppressing angiogenic activity for 1 month [68, 100]. This was confirmed by the nAMD pilot studies so monthly injections were used in both the nAMD and DME trials [17, 149]. Excellent results were achieved with monthly dosing of patients with nAMD, but attempts to extend the treatment interval to 3 months resulted in progressive loss of vision [144]. Eyes that were treated with 3 monthly injections and observed for 2 months also showed vision loss of three to four letters [153], suggesting that monthly dosing of patients with nAMD was required to achieve the best results. Therefore, the monthly treatment strategy was carried over to the treatment of DME.
Evidence supporting the use of anti-VEGF therapy for DME emerged from several sources. Elevated concentrations of both VEGF and VEGFR2 were found in animal models of diabetic retinopathy [75]. Exogenous administration of VEGF into monkey eyes produces retinal changes similar to those of diabetic retinopathy, with retinal hemorrhages, increased capillary permeability, and neovascularization [147, 171]. The discovery that these changes could be prevented by the coadministration of VEGF-Trap A40 [142] was important to drug development.
Aqueous VEGF concentrations are higher in patients with DME than in those with nAMD and vein occlusions [64], and VEGF concentrations are higher in patients with severe as opposed to mild DME [65]. In eyes with PDR, vitreous VEGF concentrations fall after successful panretinal photocoagulation [5]. Retinal hypoxia may be the most potent stimulus for VEGF production in diabetes, and treating patients with continuous oxygen supplementation for 3 months significantly reduces DME [132].
4.3.3.1 Ranibizumab Clinical Trials
The important ranibizumab diabetic retinopathy trials and their key findings are listed in Table 4.5.
Table 4.5
Important diabetic retinopathy trials with ranibizumab and details their key findings
Important diabetic macular edema trials with ranibizumab (ran) | ||
|---|---|---|
Trial and phase | Cohorts | Key findings |
READ-2 Phase II 126 patients 6 months | Treatment arms: RAN RAN + laser Laser | RAN injections at baseline, 1, 3, 5 months At 6 months 1. Mean ∆BCVA were +7, +4, 0 letters 2. Mean changes in central retinal thickness were −95 μm, −82 μm, and −117 μm At 24 months 1. Mean ∆BCVA were +8, +7, +5 letters (laser group eligible for q3month RAN at 6 months) |
RISE and RIDE Phase III 759 patients Registration trials | Treatment arms: Monthly RAN 0.3 Monthly RAN 0.5 Sham/laser | At 24 months: 1. More RAN patients had three-line improvement in BCVA (34% – 46% vs. 12% – 18%) 2. RAN patients had greater improvements in mean BCVA (+11 to +13 vs. +0.5 to +3 letters) 3. RAN patients had greater improvements in macular thickness (−250 μm to −270 μm vs. −125 μm to −133 μm) 4. RAN patients had significant improvements in diabetic retinopathy severity scores. At 36 months: 1. No significant changes in BCVA or macular thickness in RAN arms. 2. Sham/laser crossed over to RAN and improved by +2 letters |
DRCR.net Protocol I Phase III 854 patients | Treatment arms: RAN + laser RAN + def laser IVT + laser Laser | At 12 months: 1. Improvements in mean BCVA were +9, +9, +4, and +3 letters. 2. BCVA improvements in IVT + laser group that were pseudophakic at baseline were similar to RAN arms. At 36 months: 1. Patients treated with RAN + def laser improved by 2.9 letters more than RAN + laser 2. Compared to patients in RAN + laser, patients in RAN + def laser received three more injections but three fewer lasers |
RESOLVE Phase II 152 patients | Treatment arms: RAN 0.3 RAN 0.5 Sham/laser RAN groups were eligible for dose doubling | At 12 months: 1. Mean BCVA improved by +10.3 letters in pooled RAN groups vs. −1 in sham/laser group 2. 2-line and 3-line improvements in 60.8 and 33% of RAN patients and 18.4 and 5% of sham patients 3. 68% of RAN patients required higher drug dose at some point |
RESTORE Phase III 345 patients | Treatment arms: RAN RAN + laser Laser | At 12 months: 1. Mean BCVA improvements were +6.1, +5.9, and +0.8 letters. 2. Mean improvements in macular thickness were −118.7 μm, −128.3 μm, and −61.3 μm At 36 months: 1. BCVA in laser arm caught up to RAN + laser after crossover to RAN at 12 months |
Human nAMD trials were evaluating ranibizumab well before pilot studies tested its use in patients with DME. The efficacy and safety of ranibizumab in DME proceeded along three distinct clinical lines: Diabetic Retinopathy Clinical Research Network (DRCR.net), READ/RISE/RIDE, and RESOLVE/RESTORE. The US National Eye Institute, which funded DRCR.net group, was the first to demonstrate the superiority of ranibizumab with immediate or deferred laser over standard of care (laser photocoagulation). The READ trials followed by RISE and RIDE were North American registration trials that led to the approval of monthly 0.3 mg ranibizumab by the US FDA. The RESOLVE/RESTORE trials were performed in the eastern hemisphere and led to the approval of 0.5 mg ranibizumab by the European Medicines Agency (EMA).
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


