Fig. 38.1
Molecular therapies with targets associated with the hallmarks of cancer . The inner yellow circle shows the hallmarks of cancer as defined by Hanahan and Weinberg. The middle purple ring contains proteins that govern these hallmarks. The outer green circle contains exemplary molecular therapies that explicitly such molecules.
The National Cancer Act of 1971 , signed by President Richard Nixon, declared “…war on cancer…” and announced full Congressional and presidential support to eradicate the disease. As a result of increased funding and technological advancements, our understanding of cancer biology on the genetic and molecular level has exploded. Today, cancer phenotypes are associated with genetic and molecular culprits along with complex networks of various regulatory mechanisms that together cause and sustain cancer. Superficially, the genes capable of inducing carcinogenesis are divided into two categories. The first is a tumor suppressor , which is a gene that if inactivated, restricts cell division. Genes that confer pro-survival changes if activated are oncogenes. Intuitively, tumor suppressors such as p53 are commonly inactivated in cancer while oncogenes such as myc are activated and/or overexpressed. These carcinogenic changes manifest themselves by a number of avenues including overexpression, mutations, deletions, loss or gain of alleles, epigenetic modifications that alter genomic structure, alternative splicing, interference with the translation and transcription of the gene, chaperone-mediated protein folding, protein degradation processes, and posttranslational modifications that modulate localization, protein-protein interactions, and/or activity of the protein. Whether some of these changes occur simultaneously, sequentially, or otherwise is highly context-dependent and debated. Furthermore, the functional consequences of these alterations have a wide range of effects through crosstalk in cell signaling pathways. In the background of cancer-associated genomic instability, these wide-spread alterations and the functional redundancy of various genes provide a breeding ground for therapeutic resistance and make targeting cancer cells at the molecular level a challenging feat.
Radiation, surgical resection, and chemotherapy still comprise the vast majority of first-line cancer therapy today. While chemotherapy has yielded enormous patient benefit it is often accompanied by side effects that limit dose and therefore efficacy. Traditional chemotherapy is based on the notion that cancer cells divide more rapidly than normal cells and consequently will be differentially affected by an inhibitor of cell division. However, many types of normal cells need to divide for normal function. Our increased understanding of cancer has yielded numerous molecular targets for cancer therapy that may be less necessary for normal cell function and therefore may be less toxic. For instance, most normal cells divide a finite number of times, which is called the Hayflick limit. When cells divide, the ends of their chromosomes termed telomeres shorten as result of the DNA replication process, a phenomenon called the end replication problem . Upon reaching a critical telomere length, cells stop dividing and enter a dormant state referred to as cellular senescence. Cancer cells must evade senescence as they need to propagate indefinitely. Cancer cells escape this phenomenon by activating telomerase, an endogenous enzyme capable of elongating telomeres. As this oncogenic process is essential to cancer cells but not essential to most types of normal cells, telomerase is an attractive target for cancer therapy. Several types of inhibitors targeting various aspects of the telomerase molecular machinery and function are being investigated as a novel cancer therapy. There have been and continue to be efforts to discover therapies that alter the function of cancer-specific targets such as this. The rise of targeted therapies over the past two decades is a result of the rich marriage of our modern tools to understand cancer and our ancient desire to cure it. This chapter details the discovery, translation, development, and exemplified therapeutic concepts of several novel cancer therapies that specifically target oncoproteins and have molded how we discover and develop novel therapies today (Fig. 38.2).
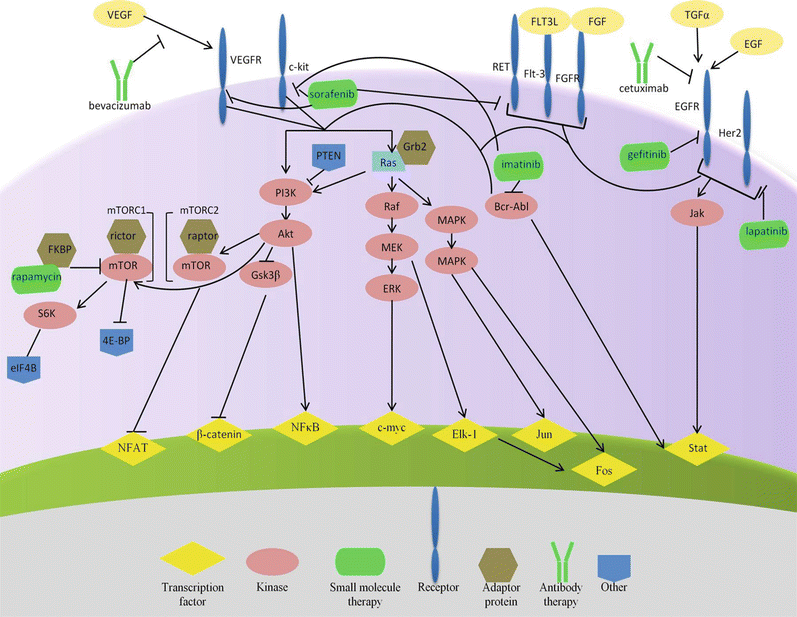
Fig. 38.2
Outline of exemplary molecular therapies targeting oncoproteins and their representative signaling networks.
38.2 Small Molecules with Big Consequences
A narrow library of atomic arrangements make up the relatively small number of molecular building blocks of cellular life such as nucleic acids that comprise DNA and RNA as well as amino acids that the end product, proteins, are made of. These building blocks are themselves synthesized from other molecules during cellular uptake and metabolism. Life has evolved to find chemical means for cells to convert small molecules to other molecules that are more useful for them. In a sense, we attempt to do the same for cancer therapy through medicinal chemistry to make more effective therapies from lead compounds. Small molecules offer a vast range of activity: sucrose sweetens foods and beverages, sodium pentothal is lethal, and amoxicillin can cure many bacterial infections. Our expanding knowledge in chemistry enables us to modify small molecules in nature or our synthetic libraries and create new molecules with altered functions, just as a cell does. Our understanding and ability to produce biological molecules on a therapeutic scale has been a relatively recent endeavor, therefore chemotherapies are almost entirely composed of small molecules. The availability, diversity, synthetic amenability, cost, and size of synthetic and natural compounds make small molecules an irreplaceable source of therapies.
38.2.1 A Rational Success Story: Imatinib
Chronic myelogenous leukemia (CML) is a cancer that causes increased amounts of white blood cells and is almost always associated with a specific translocation of chromosomes 9 and 22. The resulting shorter chromosome 22 is known as the Philadelphia chromosome, a tribute to the city housing the researchers who identified this in 1960s. The Philadelphia chromosome encodes a fusion of the two genes that results in the production Bcr-Abl. Abl is a tyrosine kinase that becomes constitutively active in the gene fusion product. This unregulated kinase activity causes oncogenic cell signaling shown to be sufficient to induce leukemia in mice. As a poster-child for translating modern cancer knowledge to clinical benefit, imatinib has significantly improved CML patient response rates. The FDA approved imatinib in 2001 for the treatment of CML as the first cancer therapy to target an intracellular molecule. Imatinib is a direct result of medicinal chemistry performed on a molecule identified as a protein kinase C (PKC) inhibitor [4]. Chemical modification of the PKC inhibitor altered the specificity of the molecule and rendered the derivative a potent inhibitor of v-abl [5], c-kit, and platelet-derived growth factor receptor (PDGFR) [6]. All of these proteins are receptor tyrosine kinases (RTKs) that bind extracellular factors and transduce the signal by phosphorylating specific protein substrates. The observation that imatinib inhibits Abl and BCR-Abl led to preclinical development and ultimately clinical trials of imatinib as a cancer therapy for CML. The concept of taking the currently available knowledge of a particular target and identifying a way to alter its function for therapeutic benefit is known as rational drug design.
Structural biology has played a key role in understanding how molecules look, how they move, how they interact with other molecules, and how all of these things change in different environments. Computational molecular docking in concert with an X-ray crystallographic structure of the catalytic domain of Abl bound to imatinib revealed that imatinib binds to the ATP-binding site of Abl preferentially when the protein is in the inactive form (Fig. 38.3) [7, 8]. The crystal structure also showed that a chemical group added to increase solubility also forms hydrogens bonds with two residues of Abl. The insight gained from such structures provides an understanding of how and where a drug binds, what types of interactions are formed, and what role each residue or functional group plays. Due to the relatively facile excision and crystallization of the catalytic domains of many kinases, several atomic structures of these proteins are freely available in the Protein Data Bank (PDB) . This provides fertile ground for numerous applications of computational chemistry to foster drug discovery and development. In addition to structural biology, medicinal chemistry also provides insight into the function of particular atoms of a molecule while searching for a more therapeutically potent derivative. The drug development process typically begins by identification of lead molecules using various screening techniques that search for a desired effect. Validation of these leads and optimization by medicinal chemistry ensues to identify the most promising compound to continue to develop. The process of lead optimization and elucidative structural biology intrinsically discerns the role of particular parts of drug and target molecules in the activity of the drug, giving rise to structure–activity relationships (SARs).
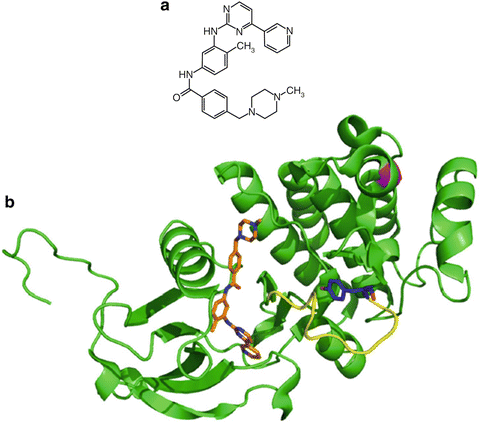
Fig. 38.3
Crystal structure of imatinib bound to Abl kinase. The Abl kinase (green) is represented as a secondary structure cartoon with its activation loop highlighted in yellow. The confirmation is in an inactive kinase state. PBD accession 1IEP.
All of the kinases in the human genome, called the kinome, share a high degree of sequence and structural homology. This means that identifying inhibitors that are specific for a given kinase is challenging and often kinase inhibitors have multiple targets. Imatinib is no exception, as it inhibits autophosphorylation of c-KIT, platelet-derived growth factor (PDGF), and ARG [9] kinases in addition to BCR-ABL. c-KIT is a receptor tyrosine kinase that is almost ubiquitously mutated in gastrointestinal tumors (GISTs) during the transformation of interstitial cells of Cajal located in the gastrointestinal tract. This mutation typically occurs in exon 11 of the c-Kit gene, which results in constitutive autophosphorylation of the protein that continually activates downstream pro-survival signaling that is oncogenic. The observation that imatinib inhibits c-KIT lead to preclinical development and clinical trials with the treatment of GISTs. Seven years after its approval for treatment of advanced CML, the FDA approved imatinib for treatment of GISTs following surgical removal of the tumor.
This extension of clinical applications demonstrates a few key concepts in cancer therapy. Firstly, exclusive specificity of a cancer drug for a molecular target is a virtue rooted in our movement toward targeted therapy. We know that without specificity, effects on normal cells can yield side effects that are deleterious. However the distinction should be made that while cancer therapies should target cancer-specific properties, this does not have to be accomplished by targeting a single molecule. It is conceivable that evolving therapeutic resistance is easier against a single target rather than an array of molecules. Furthermore, different types of tumors seem to rely on alterations in multiple genes and so multiple targets may allow for broader spectrum and more potent antitumor activity. The clinical extension of imatinib to GISTs also underscores the importance of identifying and understanding the molecular targets of therapies and how they fit with our molecular understanding of cancer. The application of a therapy used in distantly related clinical settings is a concept that continues today. Such applications are often a direct result of rational drug design and typically have an expedited timeline for starting clinical trials as they have already been tested in humans. The time and cost associated with development of cancer therapies is astounding, taking an average of over 14 years and $2 billion to reach FDA approval of a cancer drug with a success rate of just over 7 % [10]. Clearly, reducing the time spent in early phase trials profiling safety of the drug would reduce cost and expedite evaluation and patient benefit.
Therapeutic resistance is frequent in cancer. Cancer is defined by its uncontrollable cellular division and therefore is a disease of evolution governed by natural selection. While our cells copy our genome during cell division with amazing fidelity, the molecular machinery that performs this task is not completely error free. This endogenous source of mutations, environmental mutagens such as UV radiation or tobacco, and the genomic instability associated with cancer provide a sufficient source of heritable variability. Cancer treatments serve as a selective force for the cancer cells. Imagine yourself looking at population of millions of CML cells circulating in the blood that have the oncogenic Bcr-Abl fusion gene. Now introduce imatinib into the blood stream, which inhibits Bcr-Abl, and watch as the CML cells begin to die due to their dependency on the function of this oncogenic protein. If any single cell out of these millions of CML cells evolved into cancer by a process not involving BCR-ABL or can continue to divide using other oncogenic alterations, the cell will survive and continue to divide. The entire offspring of such cells will not rely on BCR-ABL to propagate and thus the patient will now not respond to therapies that target BCR-ABL. The enhanced sources of genetic variability and the solitary goal to divide more rapidly give rise to heterogeneity of tumors. This heterogeneity has significant implications as to how cancer is diagnosed and managed.
There are several ways that cancer cells get around therapeutic road blocks in intracellular signaling to keep propagating. In the case of imatinib, mechanisms of resistance include increasing the amount of Bcr-Abl to saturate the drug, mutating Bcr-Abl to still be constitutively active in the presence of imatinib, bypassing BCr-Abl and activating its downstream targets to achieve the same end goal, or simply getting rid of the drug altogether. Following the first route, cell culture and patient data revealed that overexpression of Bcr-Abl by gene duplication occurred in refractory patients and that this was sufficient for imatinib resistance [11–14]. Additionally, several patients were found to have mutations in the BCR-ABL that allowed for sustained signaling in the presence of imatinib. Due to the availability of the drug-bound crystal structure of the Abl catalytic domain, the effects of these point mutations were rationalized at the molecular level. Another intriguing avenue of drug resistance is getting rid of drugs by upregulating molecular efflux pumps localized at the cell membrane. The most characterized member of this family of proteins that induce multidrug resistance is p-glycoprotein (PgP), exhibiting broad substrate specificity to include several chemotherapies such as vinblastine, doxorubicin, and paclitaxel. Upregulation of PgP was also found in imatinib-resistant clones and in advanced CML patients. Pharmacological inhibition of multidrug resistance proteins is being explored in clinical trials in combination with chemotherapy with mixed success [15–19]. Strategies to circumvent imatinib resistance in the clinic include increasing the dose, changing to other investigational therapies, and administering alternative Bcr-Abl inhibitors such as nilotinib and dasatinib. Nilotinib and particularly dasatinib also target Sarc-family kinase (Src) and have significantly improved patient outcome after imatinib failure [20–29]. Treating patients that are resistant to first-line therapies is a challenge in clinical oncology and is a major barrier in getting an investigational drug approved as these are often tested in these refractory patients.
Ten years after the discovery of imatinib, it was approved in the US, Japan, and Europe as the first-line therapy for CML. The story of imatinib is a testament to our modern molecular understanding of cancer. Computational biologists, chemists, cell biologists, translational oncology researchers, and clinical oncologists from various countries around the world collaborated to discover and translate this therapy. Such interdisciplinary integration is a recipe for success in modern drug discovery and development and is a theme found throughout molecular targeted cancer therapy and beyond.
38.2.2 Gefitinib
Growth factor signaling is intimately involved in tumorigenesis and propagation and is involved in two of the hallmarks of cancer. Growth factor signaling typically involves the binding of an extracellular factor by a transmembrane receptor on the cell surface, which triggers intracellular signaling events. These events such as substrate phosphorylation ultimately allow for cellular proliferation through various mechanisms such as turning on transcription factors that activate genes necessary for cell cycle progression. One group of receptors that mediate several of such signals is the epidermal growth factor receptor (EGFR) family and as a consequence, this family is commonly altered by mutations and/or overexpression in a wide range of solid tumors. These receptors form homo- or hetero-oligomers upon binding various ligands to trigger a range of intracellular signaling events via Ras/Raf/MAPK, PI3K/Akt, STAT, or Src kinase pathways. EGFR is one member of this family that homodimerizes upon binding and induces proliferation through ERK, PI3K/Akt, Ras, and STAT signaling pathways. Due to the frequency of their alteration in a variety of cancers and its plethora of potent downstream oncogenic targets, the EGFR family members have successfully targeted by a number of therapeutic approaches over the past two decades.
Gefitinib is an orally active EGFR inhibitor identified by Astra Zeneca and first reported in 1996. A quinazoline derivative was found to be an ATP-competitive inhibitor and highly specific to EGFR over its related family members. Cell-based events in accordance with the inhibition of EGFR activation were observed such as autophosphorylation, upregulation of the CDK inhibitor p27Kip1, and transcriptional inhibition of the transcription factor c-Fos [30, 31]. Preclinical studies found that gefitinib had cooperative to synergistic combinations with several chemotherapies in EGFR-overexpressing cancer cell lines [32]. Interestingly, another group reported similar effects with gefitinib-chemotherapy combinations but in cancer cell lines with low EGFR expression [33]. Oral and intravenous administration of gefitinib in rats and dogs found the bioavailability of the drug to be ~50 % and that the drug was well distributed throughout the body [34]. Pharmacokinetic (PK) studies indicated that oral administration of gefitinib at 100–700 mg/day was well tolerated, had a terminal half-life between 1 and 2 days, and reached serum concentrations that inhibited 90 % of EGFR activity in vitro [35–39]. The first phase I trial of gefitinib was a dose escalation study in patients with various solid tumors that reported objective partial responses in NSCLC patients at oral doses ranging 300–700 mg per day on a 14 days-on, 14 days-off schedule [40]. The observed dose range with antitumor activity was below the dose-limiting toxicity (DLT) reported and was corroborated with another phase I trial [41]. These clinical trials used high-performance liquid chromatography (HPLC) with mass spectrometry (MS) to monitor serum concentrations of gefitinib [42].
HPLC along with complementary molecular identification techniques are often employed in clinical trials involving small molecules. HPLC allows for the separation and quantification of molecules based on their absorbance and interactions with a solid matrix. Molecular properties such as size and charge along with instrument and solvent parameters determine how the molecule interacts with this matrix. These interactions determine how long the molecule takes to migrate through the matrix column. HPLC conditions are optimized to allow for quantitative identification of a given molecule based on this empirically determined elution time, called retention time. The calculated serum concentrations can then be used to determine a plethora of pharmacokinetic parameters. These parameters are particularly important in guiding dosing schedules of new therapies and rationalizing patient responses. This technique is often coupled with mass spectrometry to allow for further verification that the molecule identified at a particular retention time is indeed the target molecule. Mass spectrometry is an electromagnetic separation method that distinguishes molecules based on their size to charge ratio of ionized forms of the molecule, which are generated by molecular collisions. Together, these ionized fragments yield a unique fingerprint for each molecule. Coupling HPLC and mass spectrometry has been instrumental in increasing the accuracy and reliability of pharmacokinetic data and identifying metabolites of drugs. Direct and indirect modifications of drugs often occur once delivered due to the staggeringly diverse mixture of molecules presence in the blood, digestive system, etc. As for gefitinib, HPLC-MS identified desmethyl-gefitinib as a metabolite of gefitinib that is inactive in vitro and in vivo [43]. Identification of metabolites is important for understanding and monitoring the various molecular species responsible for therapeutic activity as well as guiding chemical optimization.
Phase II trials with gefitinib again yielded some patient benefit in NSCLC along with low toxicity [44, 45]. With data available from phase III clinical trials, the FDA granted accelerated approval for gefitinib as a third-line therapy in NSCLC in 2003. This type of approval is granted based on promising clinical evidence of efficacy when there is no current therapy for a particular clinical setting. However, this approval is temporary and full approval is contingent on a more complete clinical data set. In the few years following accelerated approval of gefitinib, several studies found responses in the overall population of NSCLC patients [44–46]. However, a small subset of responders was identified amongst these trials with the following characteristics: female, Asian, never-smokers, adenocarcinoma, and mutant EGFR [46–48]. Several other studies confirmed efficacy of gefitinib in NSCLC patients with EGFR mutations [49–52] though no benefit was found in NSCLC patients with EGFR gene amplification [53]. A breakthrough study published in 2004 found that the majority of gefitinib responders had EGFR deletions or point mutations clustered at the ATP-binding site of EGFR, which results in a ten-fold increase in sensitivity to gefitinib [54]. Others studies corroborated these EGFR alterations in gefitinib-responsive patients [55, 56]. These genetic alterations were structurally modeled and rationalized using the crystal structure of the human EGFR kinase domain bound to gefitinib (Fig. 38.4) [57]. These response-determining mutations occur at the active site of the kinase domain in structural regions that are responsible for autoregulation of kinase activity. Further studies found that these particular mutations in EGFR stabilize the active form of the kinase and shift its affinity from ATP toward gefitinib [58].
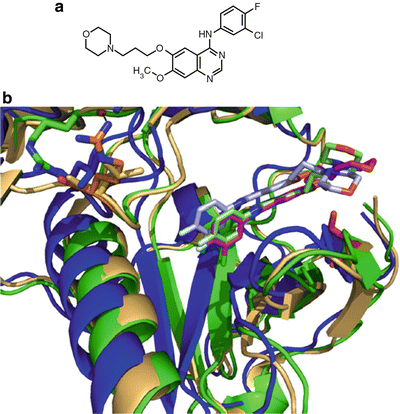
Fig. 38.4
Molecular structure of gefitinib and gefitinib bound to EGFR. (a) Molecular structure of gefitinib. (b) Crystal structure of gefitinib bound to EGFR kinase domain. Overlay of wild-type (blue), L858R (green), or G719S (beige) EGFR bound to gefitinib shown in gray, light green, and magenta, respectively. The L858R point mutation is shown in orange and its hydrogen as a black dashed line while the G719S point mutation is shown in red. PBD accessions 2ITY, 2ITZ, 2ITO.
One study mandated by the accelerated FDA approval of gefitinib and another phase II clinical trial found no benefit with gefitinib in NSCLC refractory to first-line or second-line therapies. This led the FDA to restrict its usage to patients who have previously benefited or are currently benefiting from gefitinib in 2005. Subsequent clinical trials have supported gefitinib following chemotherapy resistance [59, 60]. Together, the clinical trials comparing gefitinib and chemotherapy are confusing as they conclude significant to no benefit of gefitinib over chemotherapy. These conflicting results have been ascribed to fundamental differences in the therapeutic mechanisms of chemotherapy and targeted therapy, differences in patient populations, and biomarker selection and technique accuracy [61]. Recent studies have supported gefitinib as a first-line therapy due to demonstrated superiority over standard of care therapy in mutant EGFR NSCLC patients [62–64]. This data is anticipated to extend the restricted use label of gefitinib to its use as a first-line therapy in NSCLC patients with mutant EGFR. The use of targeted therapy based on molecular determinants surfaces later in the chapter and is increasingly integrated into FDA approval stipulations as personalized medicine emerges in practice.
38.2.3 Two Birds with One Stone: Iapatinib
As discussed elsewhere, the EGFR family is overexpressed in numerous cancers. One particular EGFR member, Her2, does not bind ligands but can form heterodimers with other ligand-bound family members to transduce oncogenic signaling through the Ras/Raf/MAPK cascade. By 2001, several recent EGFR family-targeted molecular therapies had demonstrated significant clinical efficacy in cancer including the EGFR-targeted therapies gefitinib, erlotinib, and cetuximab along with anti-Her2 monoclonal antibody herceptin. Her2 and EGFR are well-established therapeutic cancer targets based on these clinical successes, the prevalence of EGFR family alternations in cancer, and the highly homologous and druggable ATP-binding site shared by EGFR family members. A collaboration of private companies including GlaxoSmithKline launched a large synthetic effort to simultaneously target these two proteins [65–67]. Published results of these efforts detail the synthesis, specificity, and biological activity of several quinazoline or pyridopyrimidine compounds. One initial compound, GW2974, produced tumor stasis given orally at a dose of 30 mg/kg in squamous cell head and neck carcinoma and breast cancer xenografts. A later report found yet another compound, GW572016, to be a potent, reversible inhibitor of EGFR and Her2 even in the presence of excess EGF [68].
The full length 185 kDa Her2 protein can be proteolytically cleaved to shed its extracellular domain and give rise to its truncated form, p95Her2. p95Her2 is constitutively active which causes autophosphorylation, has a high oncogenic transformation ability, and correlates with lymph-node positive metastasis and poor therapeutic response [69–76]. p95Her2 was found to be insensitive to trastuzumab, preferentially dimerize with ErbB3, and be regulated by the ErbB3 ligand heregulin [77]. Lapatinib blocked baseline autophosphorylation and downstream signaling events induced by p95Her2. Her2 was found to contribute to androgen receptor transcriptional activity [78, 79]. Accordingly, lapatinib cooperated with the small molecule estrogen receptor-agonist tamoxifen to reduce estragon receptor-dependent transcriptional activity and inhibit the growth of a tamoxifen-resistant xenograft [80]. Lapatinib-resistance clones were generated by chronic exposure of cancer cell lines and were enriched in androgen receptor signaling events, suggesting the importance of this signaling in lapatinib response [81]. Combining lapatinib with anti-Her2 antibodies such as trastuzumab enhanced downregulation of the anti-apoptotic protein survivin and apoptosis in Her2-overexpressing breast cancer cells [82, 83] and trastuzumab-resistant cells [84]. These preclinical observations served as the basis for combining lapatinib with hormone therapy and trastuzumab.
Phase I studies in healthy volunteers found orally administered lapatinib to reach peak serum concentrations at 3 h and achieve steady state concentrations after 1 week [85]. High fat content of the patient significantly increased bioavailability of the drug, highlighting yet another clinical variable impacting therapeutic response [86–88]. EGFR-overexpressing and Her2-overexpressing metastatic carcinoma patients receiving lapatinib showed significant clinical responses and tolerated oral daily doses up to 1600 mg [89]. Lapatinib was also safe and effective as a first-line monotherapy in Her2-amplified advanced or metastatic breast cancer [90]. The agent has shown preliminary efficacy in head and neck squamous cell carcinoma [91, 92], but not in NSCLC [93] or prostate cancer [94].
Lapatinib has been combined with a variety of chemotherapies, hormone agonists, and trastuzumab. Addition of lapatinib to the FOLFOX4 or FOLFIRI chemotherapy regimen was safe [95, 96] and is now being explored for efficacy. Based on the clinically active combination of trastuzumab with capecitabine, studies with this combination were conducted on Her2-overexpressing advanced or metastatic breast cancer patients [97, 98]. The study found that the time to progression doubled when lapatinib was added to capecitabine without significant additional toxicity. Efficacy within these Her2+ patients was not limited to a subgroup [99], but a separate study found lapatinib to be effective in Her2+ but not EGFR+/Her2– inflammatory breast cancer patients [100]. This suggests that Her2 inhibition is a key mediator of antitumor efficacy in this malignancy. However, a higher lapatinib was recently found in preclinical models to have EGFR-independent and Her2-independent effects on death receptor upregulation that enhances efficacy when combined with TRAIL and TRAIL-receptor antibodies that bind to these receptors [101]. This rationalizes the clinical exploration of higher doses of lapatinib to gain increased efficacy via off-target mechanisms.
In 2007, the FDA approved trastuzumab and chemotherapy-resistant, for the treatment of EGFR+ advanced or metastatic breast cancer. An increase in progression-free survival (PFS) from 3 to 8.4 months was observed with the addition of lapatinib to letrozole in estrogen receptor (ER)-positive metastatic breast cancer relative to letrozole monotherapy [102]. Based on this data, the FDA extended the indication for lapatinib to its use with letrozole in Her2+ER+ metastatic breast cancer in 2010. Addition of lapatinib to tri-weekly paclitaxel as a first-line therapy yielded a significant increase in time to progression of metastatic breast cancer patients that was restricted to the Her2+ patients [103]. A recent study has also demonstrated the safety and potential efficacy of combination with weekly paclitaxel [104]. In agreement with lapatinib and trastuzumab combinatorial preclinical data, PFS was significantly prolonged in trastuzumab-resistant Her2+ metastatic breast cancer with the combination of trastuzumab and lapatinib [105]. Lapatinib demonstrates the power of multitargeted therapies over single-targeted therapies such as trastuzumab and continues to be explored with various therapeutic combinations as a breast cancer therapy.
38.2.4 Rapamycin : From the Ground Up
The PI3K/Akt/mTOR pathway is a complex signaling network that controls cell survival, death, and division in response to a variety of stimuli such as hypoxia and growth factor deprivation. The elucidation of this pathway started in 1975, when rapamycin was first isolated as an antifungal agent from bacteria in a soil sample from the Polynesian island of Rapa Nui, where the compound got its name [106, 107]. Follow up studies in rats found that rapamycin was a potent immunosuppressant [108] but the molecule lost attention in the scientific literature. Over a decade later, a high-profile immunosuppressant macrolide, FK506, was found to inhibit the proliferation of activated T-cells [109]. The same investigators that reported this observation also noticed structural homology between FK506 and rapamycin, which prompted a comparison study of their effects. Interestingly, rapamycin and FK506 antagonized the biological effects of each other. Experiments with radiolabelled FK506 found that rapamycin could directly compete with FK506 in cells for its undescribed binding target [110], which turned out to be FK506 binding protein (FKBP) [111–114]. However, the two molecules had different effects on signaling events involved in T-cell activation such as the transcriptional activity of NFAT or induced IL-2 transcription [115]. The structure of FKBP alone was determined by NMR [116] and in complex with FK506 by X-ray crystallography [117] which together revealed a rather unique drug binding site and revealed a strong conformational shift in FK506 and to a lesser extent in FKBP. Rapamycin was later found to bind to the same site but did not demonstrate a significant conformational shift itself (Fig. 38.5) [118].
Fig. 38.5
Rapamycin and its association with FKBP. (a) Molecular structure of rapamycin. (b) Surface representation of rapamycin (yellow) bound to FKBP (green) at a hydrophobic pocket. (c) Key interacting residues of FKBP involved in hydrophobic interactions (beige) and hydrogen bonds (blue) indicated with black dashed lines. PBD accession 1FKB.
Genetic studies in yeast found two homologous genes to be determinants of rapamycin-toxicity and as such were called targets of rapamycin (TOR1 and TOR2) [119]. Soon after, several reports identified a mammalian homologue of the proteins present in complex with FKBP that was dependent on the presence of rapamycin [120–122]. Furthermore, this mammalian target of rapamycin (mTOR) was required for the G1-arrest induced by rapamycin, which had been widely reported [110, 115, 123–126]. Rapamycin inhibits the function of the Akt-substrate mTOR [127–129], a serine/threonine kinase that phosphorylates p70S6K, which mediates growth factor signaling in response to cytokines such as interleukin-2 [125, 130–132]. mTOR was also found to impact EIF4E, a protein that inhibits translation by binding to the 5′ end of mRNA [133–136]. In the absence of rapamycin, mTOR can associated with adaptor proteins rictor or raptor to form mTORC1 or mTORC2, respectively. These complexes have different substrate specificity and cause distinct downstream signaling events.
In 1999, the FDA approved rapamycin as an immunosuppressant to prevent graft rejection in combination with cyclosporine A and steroids. However, the properties of rapamycin extend beyond this applicaiton. Phenotypic evaluation of growth inhibition in a particularly rapamycin-sensitive fungus, Candida albican, found nucleotide degradation and inhibition of synthesis as a primary mechanism of action in 1979 [137]. Nucleotide synthesis is a target of several clinically effective chemotherapies such as methotrexate and fluorouracil. This observation caught the eye of the National Cancer Institute (NCI). NCI experiments and an independent report found rapamycin to have antitumor activity comparable to that of cyclophosphamide and 5-FU in several solid malignancies and leukemia [138]. Other studies supported anticancer activity of rapamycin B-cell lymphoma [139], small cell lung cancer (SCLC) [140], rhabdomyosarcoma [141], melanoma [142], and pancreatic cancer [143]. Rapamycin was also found to inhibit angiogenesis under hypoxia [144] by causing transcriptional inhibition of VEGF [145], a process detailed later in the discussion of bevacizumab. Furthermore, rapamycin sensitized promyelocytic leukemia [141] and ovarian cancer [146] cell lines to cisplatin-induced apoptosis and inhibited transformation by PI3K or AKT. However, the clinical trial data generated in early phase trials of rapamycin as an immunosuppressant uncovered a poor pharmacokinetic profile [147, 148]. To overcome this problem, a large array of rapamycin analogues were created that are collectively called rapalogues.
Temsirolimus (CCI-779) is one of the first rapalogues and is a water soluble, chemically stable derivative of rapamycin (also called sirolimus) developed by Wyeth Pharmaceuticals. Preclinical studies found temsirolimus to have PTEN-dependent antitumor activity in a number of cancers [149–155] and like rapamycin, bound FK506bp and inhibited phosphorylation of S6K and 4EBP-1 [156, 157]. Phase I evaluation of temsirolimus found reversible mucositis or skin-related toxicity, no immunosuppressive functions, and that the major metabolite of temsirolimus was rapamycin [158]. Another study reported a linear correlation of time to progression and p70S6K kinase activity as measured in peripheral blood mononuclear cells, thus providing a pharmacodynamic marker [159]. Temsirolimus had similar toxicities and response rates at a dose of 25, 75, and 250 mg in renal cell carcinoma. A multicenter phase III study in renal cell carcinoma (RCC) demonstrated extended overall survival (OS) and PFS in patients relative to interferon A but found no support for the combination of these therapies [160]. In parallel with the publication of this study, temsirolimus became the first FDA-approved cancer therapy to explicitly target mTOR and joined sorafenib and sunitinib for the treatment of advanced RCC. A comparison of temsirolimus with other approved therapies for CML as chosen by the investigator found superiority of temsirolimus in terms of improving PFS and OS [161]. Combinations with anti-angiogenic therapies such as sorafenib, sunitinib, and bevacizumab have yielded additional toxicity and largely no benefit [162–164]. Temsirolimus anticancer activity is also being clinically explored in breast cancer [165], gynecological malignancies [166], multiple myeloma [167], glioma [168–170], small cell lung cancer [171], and neuroendocrine carcinomas.
Everolimus and ridaforolimus are two other rapalogues that are being investigated in clinical trials. Unlike temsirolimus, everolimus retains the immunosuppressive properties of rapamycin and was approved in 2010 for organ rejection prophylaxis. Clinical trials found oral everolimus to be safe at an oral dose of 10 mg/kg [172–175]. A phase III study in metastatic (RCC) patients who had failed sorafenib, sunitinib, or the combination demonstrated an increased PFS with everolimus [176]. This resulted in FDA-approval of everolimus in 2009 for this indication. Based on promising early clinical data, this agent further received accelerated approval in 2010 for subependymal giant-cell astrocytoma patients with tuberous sclerosis who are not eligible for surgery [177]. Everolimus has promising preliminary efficacy in Hodgkin’s lymphoma [178], metastatic gastric cancer [179], refractory NSCLC in combination with docetaxel [180] or gefitinib, and in breast cancer as a monotherapy [181] or in combination with letrozole [182, 183] or trastuzumab [184]. Interestingly, a phase II study with everolimus in refractory CLL reported partial responses along with an unexpected increase in the absolute lymphocytic count [185]. This has important implications for therapeutic sensitization as these cancer cells are likely to be more sensitive intravenous therapies when in circulation rather than in situ, however there is no clinical data to support this. Everolimus along with best supportive care was recently found to double PFS in patients with pancreatic neuroendocrine tumors [186, 187]. Ridaforolimus is in earlier clinical development but has demonstrated some partial responses and acceptable toxicity profiles in as a monotherapy [188–190] and in combination with capecitabine [191] and paclitaxel [192] in solid and hematological malignancies.
These three rapalogues work by virtually the same mechanism of action and have been developed separately by pharmaceutical companies. As the development of these rapalogues has happened in a relatively short time span, one is left wondering if these therapies have the same clinical efficacy. This is a difficulty inherent in the drug development process as clinical trial design dictates combinations with approved therapies. Combined with conflicting private interests, discerning the efficacy and unique roles of competing therapies with a similar mechanism of action is challenging. A recent phase II study in advanced pancreatic cancer attempted to compare temsirolimus and everolimus but toxicity and lack of objective response in any treatment group confounded this comparison [193]. Nevertheless, the discovery and development of rapamycin and rapalogues has extended the life of numerous cancer patients across several malignancies, prevent transplanted rejections, and elucidated a critical cell signaling pathway. At a conceptual level, the rapamycin story highlights the ability of existing therapies to be applied to other medicinal situations, the insight that can be gleaned from mechanistic studies of pharmaceuticals, and the power of medicinal chemistry.
38.2.5 Sorafenib
Raf is a serine/threonine kinase that is the apical member of the MAPK signaling cascade, which mediates a variety of cellular processes such as cell death, proliferation, and differentiation in response to extracellular stimuli. Aberrant Raf is observed in about 30 % of human cancers and correlates with the progression of prostate cancer to androgen insensitivity [194]. This increased signaling can result from alterations in upstream members such as Ras or in one of the three isoforms of Raf1 (A-Raf, B-Raf, and C-Raf). The V600E mutation in B-Raf is seen commonly in melanoma and NSCLC [195]. C-Raf overexpression has been noted in hepatocellular carcinoma [196] and validated in preclinical models as a potent drug target for ovarian cancer [197]. Earlier reports found direct evidence for Raf in determining tumorigenicity and sensitivity to radiation that posed Raf as a drug target [198, 199]. Bayer Pharmaceutical and Onyx Pharmaceutical jointly performed a high-throughput screen for Raf1 inhibitors. The screen found 3-thienyl as a lead compound with a Raf1 IC50 of 17 μM and that adding a methyl group at a particular position results in a tenfold decrease in IC50. A follow-up screen with a library of analogues yielded 3-amino-isoxazole with an IC50 of 1.2 μM [200]. Substitution of the phenyl group of this compound for a 4-pyridyl moiety lowered the Raf1 IC50 to 230 nM as well as increased aqueous solubility. This compound was found to be orally active, inhibit signaling downstream of Ras through MEK and ERK, and inhibit cancer cell growth in vitro and in xenografts [201]. Based on SARs found throughout this process, further chemical modifications were explored and ultimately yielded sorafenib with a Raf1 IC50 of 6 nM (Fig. 38.6) [202].
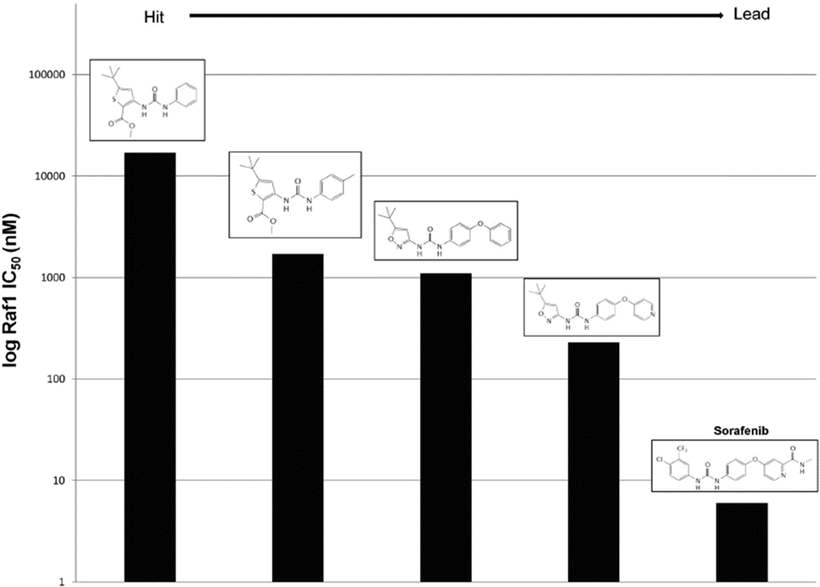
Fig. 38.6
Structure–activity relationships of Raf1 inhibitors explored during hit to lead development of sorafenib.
Sorafenib reduces MAPK signaling by inhibiting numerous oncogenic kinases: wild-type and V600E B-Raf, the angiogenic VEGFR family, platelet-derived growth factor receptor-β (PDGFRβ), fibroblast growth factor receptor 1 (FGFR1), the neurotrophin receptor RET, and the cytokine receptors c-Kit and Flt-3 [203, 204]. The growth of several xenografts were inhibited by sorafenib though in a few cases, no change in MAPK signaling was detected but was rationalized by its anti-angiogenic effects of VEGFR inhibition [203]. Crystal structures of wild-type and V600E B-Raf in complex with sorafenib revealed key interactions with residues conserved in c-Raf [205]. The pyridyl ring occupies the ATP-binding pocket while the trifluoromethyl phenyl ring occupies a proximal hydrophobic pocket. Interestingly, the nitrogen of the pyridyl group forms a hydrogen bond with B-Raf and rationalizes the potent increase in activity following substitution of the pyridyl group for the phenyl group. The urea group bridging the rings was found to form a hydrogen bond network with protein, explaining its conservation throughout the analogue search and development. In this case, the structural data rationalized the previously found SARs. However, structural data can conversely be used to guide the exploration of analogue and can also potentiate in silico screen that computationally models the binding of a virtual library of ligands to protein structure. It should be noted that crystal structures do not provide a complete platform for computing binding affinities. While the biophysical details of ligand binding are beyond the scope of this chapter, the affinity of two molecules is determined by the change in enthalpy and entropy. As crystal structures represent a single average molecular conformation across the crystal lattice, they do not reflect the motions of the molecule. Therefore crystal structures cannot provide direct entropic information themselves and by extension do not fully represent binding affinity.
Since sorafenib inhibits a multitude of kinases, preclinical and early clinical trials explored a variety of malignancies. Several phase I trials found dose-dependent responses with an optimal oral dose of 400 mg [206–208] and addition of sorafenib to a variety of standard of care chemotherapy regimens did not increase toxicity profiles [209–214]. Renal cell carcinoma (RCC) is particularly resistant to the majority of chemotherapies and until 1997 was treated with interferon that causes significant toxicities and limited responses. Due to the poor clinical outcomes with standard of care therapies, preclinical efficacy of sorafenib, and an encouraging phase I result in a metastatic RCC patient, phase II studies were enriched with RCC patients. This study found a strong response in RCC patients [215, 216] and led to a large-scale phase III study that reported a doubled PFS and ~40 % increase in OS [216, 217]. These results gained sorafenib FDA-approval at the end of 2005 for RCC. A subset analysis of this phase III trial found similar clinical benefits regardless of previous cytokine therapy [218] and a follow-up >1 year from treatment initiation found sustained efficacy and a well-tolerated toxicity profile [219]. Liver transplant has traditionally been the only treatment option available for hepatocellular carcinoma. However, many patients become ineligible while waiting for the transplant as a result of disease progression. Human xenografts of liver cancer cell lines showed partial tumor regression from sorafenib and inhibition of ERK and EIF4 [220]. A phase II trial found moderate efficacy and that time to progression correlated with phospho-ERK levels [221]. A multicenter phase III trial demonstrated an unprecedented increased OS in HCC and solicited the FDA to extend the indication of sorafenib to unresectable HCC [222].
Due to the multitargeted nature of sorafenib, it is being explored as a monotherapy and in combination with chemotherapies in a variety of malignancies, though biomarkers are difficult to find. A recent trial in metastatic melanoma found no correlation between clinical responses and BRAF V600E mutation status, cyclin D1, or the proliferation marker Ki-67 [223]. Early evidence was promising with sorafenib, carboplatin, and paclitaxel in melanoma [224] but a recent phase III study in advanced melanoma failed to demonstrate any benefit as second-line therapy [225]. A phase III trial in advanced HCC doubled PFS and OS with sorafenib plus doxorubicin compared to doxorubicin alone [226]. Other promising efficacy of sorafenib has been seen as a neoadjuvant in advanced RCC [227], as a monotherapy [228] or with erlotinib [229] or gefitinib [230] in NSCLC, metastatic RCC with gemcitabine and capectibaine [231]. Sorafenib has had limited to no clinical efficacy in malignant mesothelioma [232], prostate cancer [233, 234], uterine cancer [235, 236], sarcomas [237], advanced and metastatic squamous cell carcinoma [238], with paclitaxel and carboplatin in NSCLC [239, 240], or as a neoadjuvant in advanced ovarian cancer [241], sunitinib-refractory metastatic RCC [242].
38.2.6 Vemurafenib
Melanoma is particularly dependent on signaling through the c-Kit/NRAS/BRAF/MEK/ERK signaling axis. This solicits the clinical application of imatinib to melanoma as it inhibits c-Kit among other targets. However, targeting c-kit has been clinically limited in melanoma as several patients have gene amplification or have oncogenic alterations downstream in this signaling axis. Targeting BRAF in melanoma was explored during the clinical development of sorafenib but ultimately proved ineffective as a monoagent and did not improve OS in combination with carboplatin and paclitaxel in a phase III placebo-controlled trial [225]. Several reasons have been proposed for this failure such as unsaturated MAPK inhibition at the MTD of sorafenib [243] and furthermore, the ability sorafenib to target BRAF in vivo has been challenged [244].
The V600E activating mutation in BRAF is commonly observed in melanoma and results in resistance to therapies targeting upstream molecules. Using structural biology, vemurafenib was developed as a selective inhibitor of BRAFV600E [245, 246], though other targets have been uncovered such as CRAF, ACK1, SRMS, and MAP4K5 [247]. Phase I studies with Vemurafenib reported objective responses [248] that were corroborated in phase II studies and found pERK to be a valid correlative response marker [249]. A large phase III study in therapy-naïve patients ineligible for resection was recently reported an increased PFS and OS relative to dacarbazine [250]. In 2011, the FDA approved vemurafenib for the treatment of unresectable or metastatic melanoma with BRAFV600E.
While verumafenib is clearly a clinical oncology success, patients with wild-type BRAF still need better treatment options, biomarkers are needed to preemptively identify unresponsive patients with melanoma harboring BRAFV600E, and many patients relapse despite the improvements in OS [251]. Verumafenib-resistant melanoma cells appear to upregulate PDGFR, NRAS, or MAPK signaling in vitro and in tumors [252, 253]. Preclinical evidence suggests that targeting MEK, PI3K, and mTORC in vemurafenib-refractory patients may be an effective combinatorial strategy [254]. Other preclinical reports have suggested the combination of vemurafenib with metformin [255], immunotherapy [256], or a monoclonal antibody targeting chondroitin sulfate proteoglycan 4 [257].
38.3 Antibodies
While evolution has allowed cells to use small molecules to remain viable, it has allowed organisms to protect themselves from disease through the immune system. Antibodies are large proteins used by the immune system to recognize, respond to, and remember foreign biological material. Antibodies contain a region that is highly specific for a protein target associated with the biological material known as the hypervariable region (Fv), which allows for a high degree of diversity and specificity. The constant region (Fc) of the antibody consists of a particular immunoglobulin that determines the class of antibody and ultimately the type of immune response mounted. Several factors including the complexity of these large proteins have curtailed our ability to synthesize these highly specific binding molecules. However in 1975, medicine was forever changed by discovery the ability to harness cells to make antibodies [258]. This process consists of immunizing a mouse with the desired antigen, fusing splenic cells from the immunized mice with mouse myeloma cells using Sendai virus, selecting and growing desired clones. This capability has revolutionized modern medicine and biomedical research by offering an unparalleled ability to recognize any given protein with unparalleled specificity. Oncogene pathways mediated by soluble or cell-surface proteins naturally lend themselves as cancer therapeutic targets within the reach of antibodies.
38.3.1 Rituximab
CD20 is a B-cell-specific cell surface protein and as such, is expressed in B-cell cancers such as Non-Hodgkin’s lymphoma (NHL). While the function of CD20 remains unclear, it is known that the protein is not secreted or cleaved from the cell surface [259] nor is it internalized following antibody binding [260]. An early study found that administration of murine anti-CD20 in malignant B-cell lymphomas produced a 90 % reduction of circulating malignant cells within four hours in humans [261]. Variable regions of the murine antibody were cloned in to an expression vector to allow for an antibody with human constant regions and would later gain the name rituximab [262]. Rituximab demonstrated a binding affinity for CD20 of 5 nM and resulted in a near complete depletion of peripheral blood cells and a 40–70 % depletion of B-cells in lymph nodes that began recovery around 2 weeks after administration. How does sticking an antibody to a surface molecule that does not have an obvious functional importance potently inhibit cancer? The currently understood answer is that rituximab induces three modes of cell death mediated by immune and cancer cells (Fig. 38.7). Unlike its murine counterpart, this hybridized antibody bound C1Q in vitro and furthermore induced cell lysis in the presence of serum as a source of complement. C1Q is part of a large protein complex that is found in serum and binds IgG or IgM to trigger a series of intra-complex cleavage events that ultimately form a transmembrane complex, called the membrane attack complex, to induce osmotic lysis of the antigen-expressing cell. This process is known as compliment-dependent cytotoxicity (CDC). CDC appears to be a key aspect of the antitumor activity of rituximab as rituximab-resistant patient samples were associated with CD59, which negatively regulates this process [263, 264]. Restoration of compliment has been shown to reverse resistance in small-scale patient studies [265, 266] though this has been challenged in preclinical models [267, 268] and follicular NHL [269].
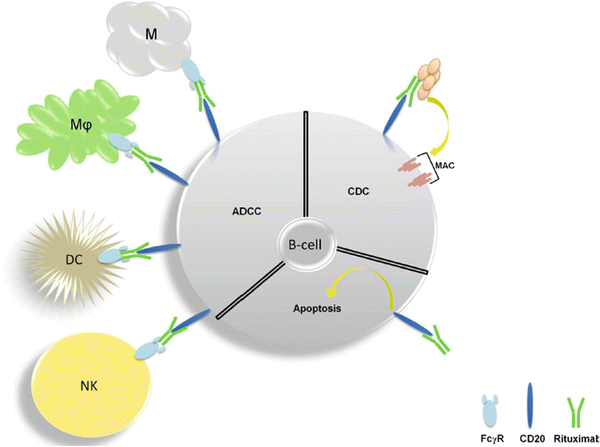
Fig. 38.7
Antitumor mechanisms of rituximab . Antibody-dependent cytotoxicity (ADCC) is mediated by natural killer cells (NK), dendritic cells (DC), macrophages (Mφ), and mast cells (M). Complement-dependent cytoxicity (CDC) involves a series conformational changes and cleavage events upon binding IgM or IgG that ultimately leads to the formation of the membrane attack (MAC) which induces osmotic lysis. Induction of apoptosis directly by CD20 also occurs but is less clear in mechanism.
In addition to CDC, effector cells such as macrophages and natural killer cells (NK) express a family of activating and inhibitory receptors called FcγR that bind the constant region (Fc) of IgG. A study in mice showed that blocking these various receptors mediated anti-CD20 mAb-induced B-cell depletion and was isotype-specific [270]. FcγR bound to IgG present on the surface of a cell can result in phagocytosis by effector cells but these events are determined by the affinity and balance of activating and inhibitory FcγR molecules [271–273]. Accordingly, a follicular NHL study found significantly higher rituximab responses in patients that harbor the FcγR 158V allotype compared to the 158F allotype, which has a relatively weaker affinity for IgG1 [274]. Ex vivo studies demonstrated that rituximab causes NK-mediated cell lysis [275–277] in a dose-dependent manner that was determined by the FcγR allotype [275]. There is also evidence that CD20 has direct effects. Early studies found that antibodies against CD20 significantly mediated RNA synthesis and cell cycle progression [278–280]. More recent evidence has found that cross-linking CD20 with antibodies in some, but not all, B-cell lines causes caspase-mediated, Bcl-2-independent apoptosis, and effects the tyrosine kinases Src, Jnk, and p38 [281–284]. The contribution of these different mechanisms appears to be highly context dependent and is likely to be a dynamic process varying between patients or even within a single patient.
Early clinical data found rituximab to have an average half-life of 18.5 days [285] and similar efficacy and favorable toxicity in relapsed NHL patients relative to CHOP chemotherapy (cyclophosphamide, doxorubicin, vincristine and prednisone) [285–289]. A small-scale study of rituximab found antitumor activity concentrated in the follicular subtype of NHL, though the population size prevented any significant conclusion [290]. Other studies corroborated the significant monotherapy efficacy of rituximab in follicular lymphoma [291, 292]. Adding rituximab to chemotherapy yielded an additive benefit and did not augment the toxicity of standard chemotherapy for NHL [293, 294]. Furthermore, polymerase chain reaction (PCR) of NHL patients with follicular histology treated with this combination showed a depletion of the chromosome 14 and 18 translocation often associated with follicular NHL [295, 296]. Another study found the disappearance of this translocation a year after a 4-week course of rituximab [292]. This translocation induces a sustained transcriptional upregulation of Bcl-2, a protein that prevents mitochondria-mediated apoptosis carried out by many tumor suppressors and inhibited by oncogenes. In 1996, rituximab became the first antibody approved by the FDA as a cancer therapy and was indicated for relapsed or refractory low-grade or follicular, CD20+, B-cell NHL. Three phase III studies (E4494, GELA, and MiNT) that were highly enriched in therapy-naïve, large diffuse cell NHL patients found a significant increase in OS at a 2 year follow-up with the addition of rituximab to CHOP or other anthracycline-based chemotherapies. Analysis of biopsies from E4494 patients found p21 expression as a rituximab-specific, independent predictor of clinical outcome [297].
NHL patients who previously received at least a single 4-week course of rituximab therapy had equivalent efficacy and toxicity after an additional course with a median internal of 14.5 months [298]. The combination of rituximab and interferon yielded no significant additional benefit in the short term in follicular NHL [299]. Today, rituximab is also approved as a first-line therapy for low-grade or follicular cell CD20+ NHL with CHOP and large diffuse B-cell CD20+ NHL with CVP (cyclophosphamide, vincristine, and prednisone). Initial studies of rituximab were conducted with intravenous administration for a 4-week cycle. However, exploration of alternative dosing schedules has provided efficacy in small lymphocytic lymphoma [300] (SLL) and chronic lymphocytic leukemia (CLL) patients [300, 301].
The addition of rituximab to fludarabine and cyclophosphamide in CLL was found to be safe [302–307] and result in unprecedented clinical responses in CLL patients [305]. A phase III study found that this combination increased the amount of patients without disease progression (65 to 45 %) and overall survival (87 to 83 %) [308], resulting in the extension of its indication to this malignancy in 2010. Shortly after, it was also approved as a maintenance therapy following a response with chemotherapy and rituximab in CD20+ NHL based on phase III evidence of prolonged PFS with 1 weekly dose [309]. Rituximab and the proteasome inhibitor bortezomib have been shown to synergistically induce apoptosis in preclinical cancer models [310, 311] and a phase II supported the combination but noted significant neurological toxicity [312]. Also targeting CD20, the monoclonal antibody ocrelizumab was approved in 2009 for CLL refractory to fludarabine and the anti-CD52 antibody alemtuzumab. Other anti-CD20 antibodies are in clinical development for lymphoma including ocrelizumab, veltuzumab, AME-133V, PRO131921, GA101. As seen with rapamycin, the ongoing elucidation of the mechanism of action of rituximab has strongly augmented our biological understanding within and beyond cancer.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


