Target Identification and Drug Discovery
Shannon Decker
Melinda Hollingshead
Robert Shoemaker
Edward A. Sausville
This chapter provides an overview of three stages of the preclinical development of an anticancer drug. The first stage is the identification of a drug target. Numerous approaches for target identification exist ranging from biologically based ones to newer chemogenomics- and RNA interference-based techniques. The second stage is the discovery of an active drug candidate with therapeutic potential and the selection of the optimal candidate or limited set of candidates for further evaluation. These studies would also ideally allow an assessment of how the new molecule differs from currently available therapeutic agents. In the third stage, early preclinical development is directed at increasing confidence that the drug lead will actually function as a useful therapeutic agent in humans. These studies focus predominantly on eliciting activity in animal models of cancer and ideally correlate the degree of antitumor activity with the pharmacology of the drug.
Cancer Drug Target Selection
Within the past 15 years, the approach to the discovery and development of cancer drugs has undergone a marked change, from focusing classically on empirical antiproliferative activity as a basis for initial interest in a compound, to selecting drug candidates on the basis of their capacity to modulate molecular targets that are important in cancer pathophysiology. Modern approaches to identifying targets include specific departure from our understanding of cancer cell biology, efforts to “retrofit” active molecules for which target knowledge is uncertain, and approaches derived from newer techniques such as chemogenomics and RNA interference.
Biologically Based Approaches
Our current view of the cancer cell leads to the view that by the time a cancer is clinically manifest in a patient, a number of genetic lesions have occurred in the tumor, resulting in discrete sets of abnormalities that may differ in detail from tumor to tumor, but exist as categories of molecular defects common to essentially all tumor types. This idea, articulated elegantly by Hanahan and Weinberg,1 points to deregulated proliferation-control pathways, loss of tumor suppressor gene function, loss of functions that would promote tumor cell programmed death (apoptosis), acquisition of limitless replication potential through telomere-replicating strategies, activation of host angiogenesis, and the capacity to invade into normal stroma as attributes of all tumors. To these features must be added the capacity of tumors to thwart the host immune system (e.g., Uttenhoeve et al.2). Of special importance to cancer drug discovery is the existence of different degrees of deregulated DNA repair processes, perhaps as a way of tolerating mutational events in the life history of a particular tumor. The result is a potential vulnerability to DNA-damaging agents. This is exemplified by increased susceptibility of cells with lesions in the ERCC1 (excision repair nucleus) to platinum.3 Each of the specific molecules or pathways creating the altered cellular state that underlies tumor cell biology as compared to the normal cell could conceivably be a target for cancer drug discovery and development.
Those molecules consistently mutated in the course of the development of cancer in patients can be thought of as defined by nature as important in the pathophysiology of that tumor and can be characterized as pathogenic targets. These molecules may come to attention not only by point mutations in their coding sequence but also by their proximity to frequently observed chromosomal breaks points or regions of DNA amplification in tumors. Examples of molecules of this type that have been already validated as cancer drug targets include the p210bcr/abl oncoprotein of chronic myelogenous leukemia4 and the HER-2-neu tyrosine kinase5 that is frequently detected by genomic amplification in breast carcinoma. Approved drugs directed at these targets include imatinib and the monoclonal antibody trastuzumab, respectively. In validation of this way of thinking, patients enjoying the best clinical response to the epidermal growth factor receptor tyrosine kinase inhibitors erlotinib and gefitinib possess a mutated, activated form of the agent’s target kinase.6, 7, 8 Alternatively, the drug target could be downstream of a mutationally activated oncoprotein. This is exemplified by the observations of Settleman et al. that among cell lines highly sensitive to inhibitors of B-raf are not only the expected B-raf mutant bearing lines but also those with amplified N-ras.9 In either event, mutational activation of a pathway defines a state of “oncogene addiction” that is the basis for strategic design of approaches to intervene pharmacologically by inhibitor design.10
Ontologic targets relate to the normal tissue of origin of the tumor. Examples of validated targets of this type include the estrogen or androgen receptors in breast or prostate cancer, respectively, and the CD20 cell surface determinant of non-Hodgkin’s lymphoma. Pharmacologic targets relate to the pharmacokinetic or pharmacology names of a drug itself. For example, dihydropyrimidine dehydrogenase is a target whose degree of activity modulates susceptibility and toxicity of fluorinated pyrimidines, or dexrazoxane modulates levels of free iron, a “target” that modulates the cardiotoxic potential of anthracyclines. Stromal or microenvironmental targets include the large array of molecules responsible for sculpting tumor stroma and supporting cell framework including mediators of angiogenesis.
The regulatory approval of bevacizumab, a monoclonal antibody to vascular endothelial growth factor (VEGF),11 presaged the intense clinical trials activities leading to the approval of the “small” molecule multitarget kinase inhibitors such as sorafenib and sunitinib that also target VEGF-R signaling. Immunologic strategies, including immunomodulating cytokines or even vaccines, might be considered as special cases ultimately directed at the tumor microenvironment.
The regulatory approval of bevacizumab, a monoclonal antibody to vascular endothelial growth factor (VEGF),11 presaged the intense clinical trials activities leading to the approval of the “small” molecule multitarget kinase inhibitors such as sorafenib and sunitinib that also target VEGF-R signaling. Immunologic strategies, including immunomodulating cytokines or even vaccines, might be considered as special cases ultimately directed at the tumor microenvironment.
Another concept that is potentially important in the identification of targets is that of “synthetic lethality,” which has been well articulated by Kaelin.12 In essence, mirroring findings from yeast genetics, two genes are “synthetic lethal” if mutation of both genes results in cell death even though mutation of either alone is compatible with cell viability. This approach envisions the identification of target genes that are synthetic lethal to a mutation relevant only to cancer cells, leaving normal cells viable. Synthetic lethal screening may also allow the identification of compounds with selective toxicity to cells bearing mutational or other silencing of genes, particularly relevant to modulators of tumor suppressor gene pathways, frequently deregulated in cancer through loss of the tumor suppressor loci. Synthetic lethal screening has become an important concept in RNA interference screening techniques discussed below.
The many approaches to target identification defined above derive from a growing understanding of cancer cell biology and have actually created a wealth of potential cancer drug targets. Indeed, extensive efforts to define potential targets with altered expression pattern in tumors have been made available through publicly funded (e.g., the Cancer Genome Anatomy Project; see http://cgap.nci. nih.gov/) and privately maintained databases. A major question is, how might these targets be prioritized?
The argument for a particular target is strengthened when a phenotype related to tumor behavior can be modulated in cell models (antisense, small interfering RNA, dominant negatives, or ribozyme approaches) or in animal models (knock-outs or transgenics) by genetic approaches to altering the presence or function of a tumor target. For example, topoisomerase IIIβ knock-out mice exhibit premature senescence, which suggests that the use of a topoisomerase poison, specific for that isoform, would drive tumor cells toward senescence.13
Other practical criteria that influence the suitability for selection of a molecular target for a drug discovery campaign include the availability (and cost) of reagents to allow screening of leads and the identification of an assay format that is amenable to high-throughput screening (HTS). Finally, the size of the patient population (market) and availability of effective treatments for a particular cancer are usually considered before investing resources toward a new target.
“Retrofitting” Active Molecules
Rather than starting from the tumor cell to find a target and a drug affecting the target, some have approached target identification by starting from or “retrofitting” active molecules. For example, geldanamycin and other benzoquinone ansamycins had been known for many years to have activity in empirical antiproliferative assays. Neckers et al. prepared a solid-phase immobilized geldanamycin derivative and used affinity precipitation in a search for molecular targets, ultimately identifying the heat-shock protein 90 (HSP90).14 Not only did this identify a target for the geldanamycins, but this work also helped spawn further drug discovery efforts for other HSP90 inhibitors. Similarly, the immunosuppressive drug rapamycin was known to interfere with the progression from G1 to S phase, but the pathway was not known. Sabers et al. used an affinity matrix of glutathione-S-transferase, rapamycin’s intracellular receptor FKBP12, and rapamycin in the isolation of mammalian targets of rapamycin (mTOR), the biology of which has now been extensively studied and exploited for drug discovery efforts.15 The fumagillin derivative TNP-470 was the subject of intense efforts to develop an effective antiangiogenic agent, although little was known of its molecular mechanism of action. Human umbilical venous endothelial cells were incubated with a fumagillin derivative complexed with biotin to test the hypothesis that fumagillin was forming an adduct with a cellular protein, from which efforts the methionyl aminopeptidases were identified as targets.16
In addition to efforts to identify targets for single active agents, computational methods have also been employed for target identification. As described later in this chapter, in the National Cancer Institute (NCI) 60 cell line screen, various pattern recognition algorithms have been used to compare the patterns of activity of known active compounds with the levels of expression of various molecular targets in the cell lines. The identified putative targets associated with a compound’s action can then serve as a starting point to deconvolute the basis for compound action for further drug discovery efforts. A more modern successor to this approach uses even larger groups (e.g., 500) of cell lines,9 but they are ones which screen for compound activity in a way that annotates target or target pathway activation status.
Newer Technologies
Newer technologies such as chemogenomics and RNA interference have also come to the forefront for target identification. These techniques also have potentially broad utility for other aspects of drug discovery and lead into the discussion of screening strategies.
Chemogenomics has been described by some as discovery and association of all possible drugs with all possible drug targets,17 involving the combination of modern genomic technologies with the effects of compounds on biological targets.18 A chemical genetics application would use small molecules to probe the functions of proteins by adding a library of small molecules to cells, selecting the molecules producing the desired phenotype and identifying the protein bound by the molecules.19 This technique can also be reversed to use the small molecules to further understand the identified protein. The Broad Institute of Harvard and MIT is home to the NCI-funded Initiative for Chemical Genetics, a public access research facility whose data utilizing several aspects of this approach are deposited into the ChemBank web site (http://chembank. broad.harvard.edu/).
RNA interference was first recognized after seeking to define mechanisms for regulating gene expression in model invertebrate systems such as worms, flies, or plants. More detailed discussions of the mechanisms related to RNA interference are summarized elsewhere.20,21 Briefly (Fig. 2-1), expression of a double-stranded “short interfering RNA” (siRNA), one strand of which has homology to a target sequence in the mRNA of a gene of interest, allows a complex to be formed with the target cellular RNA and act as a degradation mechanism through engagement of the RNA-induced
silencing complex (RISC), to result in loss of mRNAs bearing the target sequence. SiRNAs may be introduced directly into cells, or result from cleavage from “short hairpin RNAs” (shRNAs) arising from introduced plasmid DNA sequences and acted upon by cellular RNAses to cleave a spacer RNA forming the hairpin between the two stretches of complementary RNA. RNA interference technology has utility in virtually all aspects of cancer drug discovery, and is a basis for many recent innovative screening strategies, specifically reviewed in Iorns et al.22 Specific uses include target identification, synthetic lethal screening, exploration of drug sensitization or resistance mechanisms, and target validation.
silencing complex (RISC), to result in loss of mRNAs bearing the target sequence. SiRNAs may be introduced directly into cells, or result from cleavage from “short hairpin RNAs” (shRNAs) arising from introduced plasmid DNA sequences and acted upon by cellular RNAses to cleave a spacer RNA forming the hairpin between the two stretches of complementary RNA. RNA interference technology has utility in virtually all aspects of cancer drug discovery, and is a basis for many recent innovative screening strategies, specifically reviewed in Iorns et al.22 Specific uses include target identification, synthetic lethal screening, exploration of drug sensitization or resistance mechanisms, and target validation.
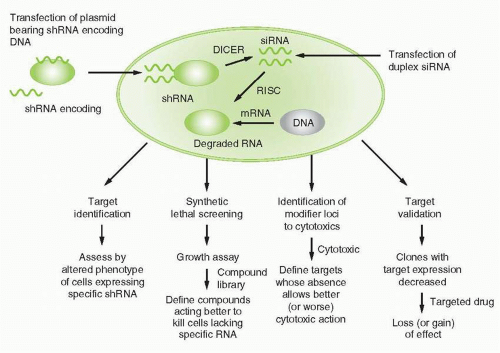 FIGURE 2-1 siRNA and its potential applications. |
For target identification, a single siRNA, or libraries of siRNAs representing a variety of potential targets, alters a cellular phenotype, for example, proliferative state, invasive capacity, survival, etc. and discloses whether the target(s) altered after expression or action of the siRNA(s) modulates the phenotype. In that event, screening strategies to define modulators of target function can then be designed. This effort can result in an assessment of targets in a particular disease context. For example, utilizing siRNA-based technology, Woo et al. identified “driver genes” in hepatocellular carcinoma, each of which might now be considered in a campaign to define hepatocellular carcinoma-related drugs.23 Specific targets relevant to the action of specific candidate drugs can also be usefully highlighted by siRNA technology. For example, Tetsu and McCormick utilized siRNAs to cyclin-dependent kinase 2 (CDK2) to reveal little impact of removing that target, and offered the point of view therefore that CDK2 might not be a suitable cancer drug target.24 This outcome actually reveals a limitation of siRNA screening, as target classes such as CDKs with redundancy of function in different family members might not be highlighted as potentially valuable target classes if only a single representative of the class is targeted. In contrast, for example, Ludwig et al. utilized siRNA-based approaches to validate that NSC73306 is a putative first in class multidrug-resistance targeted agent that actually depends for its action on the expression of mdr1.25 A particularly important aspect of this strategy is the design of control or “counterscreens” to assure that the siRNA is functioning as a specific indicator of target action across a range of cellular contexts related to eventual use of the drug candidate.
The concept of “synthetic lethality” in cancer drug screening was discussed generally above. In approaches of this type utilizing RNA interference, siRNA-mediated downmodulation of a target can result in the acquisition of sensitivity to drug action, therefore linking the action of the drug to the presence of the target. A classical demonstration of the relevance of siRNA-related synthetic lethal screens emerged from the observation that cells deficient in BRCA-1 were highly sensitive to concomitant poly(ADP-ribose) polymerase (PARP) inhibition,26 owing to the inability to repair DNA lesions utilizing homologous recombination. This logically leads to potential value of utilizing cells deficient in various DNA repair enzymes and repair-related signaling systems to identify additional determinants of sensitivity to PARP inhibitors.27,28 Other examples of this application relate to the definition of target alliance to newly defined compound action, such as the recent demonstration that apoptosis was mediated by Candida spongiolide protein kinase R function.29 In a variation of this approach, modulation of a pathway’s activation by specific siRNAs may either be mimicked by a compound or a compound class, thus identifying the compound as mechanistically involving the pathway in its action. Exemplifying this approach, Gaither et al. identified siRNAs that conveyed resistance to the “second mitochondrial-derived activator of caspases” (Smac) mimetic compound LBW242 (a putative modulator of apoptotic effect), and found that entities conveying such resistance were members of the TNF-α pathway as well as X-linked inhibitors of apoptosis (XIAP).30
Synthetic lethal screening can also focus on pathways that are modulated by the action of a siRNA or, more commonly, libraries of siRNAs. Alternatively, as also reviewed comprehensively by Gaither,18 synthetic lethal screens may provide critical information about genes or set of genes modulated by the novel compound.
SiRNA-based screening approaches can also be used to define pathway members that in a specific disease context serve as a basis for maintaining disease phenotype and therefore could serve as putative targets for drug discovery, as exemplified by modulation of the NFκB pathway in large B-cell lymphoma activated B-cell phenotype as defined by Ngo et al.31
SiRNA-based screening approaches can also be used to define pathway members that in a specific disease context serve as a basis for maintaining disease phenotype and therefore could serve as putative targets for drug discovery, as exemplified by modulation of the NFκB pathway in large B-cell lymphoma activated B-cell phenotype as defined by Ngo et al.31
Conceptually allied to synthetic lethal screening in “discovery” campaigns, siRNA-related screens have defined important aspects of drug sensitization or resistance mechanisms related to the use of established agents in a way that could lead to a more focused use of the agent. For example, Whitehurst et al. identified numerous genes apparently conveying greatly enhanced sensitivity to paclitaxel.32 Bartz et al. analogously defined genes conveying enhanced sensitivity to cisplatin, gemcitabine, and paclitaxel.33
Of critical importance to industrial prioritization in drug discovery and development campaigns is target validation, the clear demonstration that a potential target has relevance to disease mechanisms in vivo. Classical xenograft models discussed elsewhere in this chapter are frequently ambiguous in conveying enthusiasm for the development of a compound class. However, siRNA-based approaches can be utilized to create in vivo models that convey forcefully the importance of target function. For example, Hoeflich et al. demonstrated in melanoma models in mice that shRNAs to mutated B-raf,34 in this case expressed through tetracycline-responsive promoter elements, impaired growth of the model, a finding that certainly encouraged discovery of B-raf inhibitors.
Defining an Active Drug Candidate
Initial recognition of a lead compound can come from a purely molecularly targeted screen, directed against a purified enzyme or a cell engineered to overexpress or underexpress a particular target, or through a chemogenomic or siRNA-based activity described above, or from a cell- or animal-model-based antiproliferative screen, against naturally occurring or engineered tumor cells. Each screening model has its distinct advantages and disadvantages. The molecular targeted approach may yield a drug candidate with clear selectivity for a particular target, but then target modulation must be documented in a cellular context, hopefully with evidence of useful antitumor activity. Ideally, constant evidence of effects “on target” can be an important aid to lead optimization. Cell-based screens have the advantage that drug candidates defined by this route have the ability to distribute across plasma membranes and survive in the intracellular milieu. On the other hand, their mechanism of action must be determined prior to efficient lead optimization, and cell-based screens are less frequently amenable to high-throughput approaches. In contrast, pure in vitro biochemical molecular targeted screens have the attraction of being very amenable to the screening of large collections of molecules.
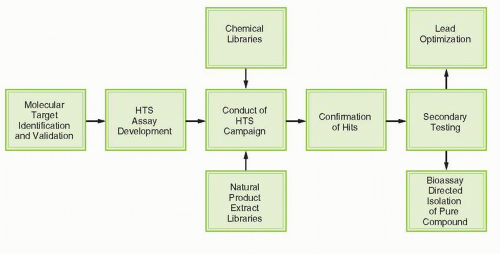 FIGURE 2-2 Generic process for high-throughput molecular targeted drug discovery. |
Figure 2-2 illustrates the generic process of high-throughput, molecular targeted screening (HTS). The process requires initial identification and validation of a target, followed by development and characterization of an assay suitable for HTS. This assay is then used to screen chemical collections or “libraries” to identify active samples that are the focus of additional testing to establish potency, selectivity, and other features important for further development. Screening is typically conducted in a “campaign” mode, with primary screening data from a particular library evaluated after the whole library has been tested. Criteria for activity are frequently established such that a “hit rate” on the order of 1% is obtained. Thus, for a library of 100,000 samples, 1,000 of the most active samples would be selected for the confirmatory testing. Efficient testing has often demanded that the primary screen is conducted at a single concentration, although some researchers are now popularizing quantitative high-throughput screening (“qHTS”) testing multiple concentration on 384- and 1,536-well plates.35 Confirmatory testing may be done using the same protocol or may be done in a more traditional concentration-response fashion to facilitate subsequent consideration of screening leads in the context of potency.
Sources of Diversity for Lead Identification
A crucial issue in any screening project for new anticancer drugs is the acquisition of compound libraries. Whether the initial screen is a target-directed biochemical screen or an empirical antiproliferation screen, the greater the structural diversity in the set of molecules examined, the more likely a novel inhibitor will be identified. Historically, natural products, defined here as extracts from plants, microbial, and animal sources, have provided an excellent source of bioactive molecules with novel structures and mechanisms
of action. In cancer treatment, natural products constitute several major therapeutic classes including the vinca alkaloids, camptothecins, and taxanes. Natural product extracts as sources for cancer drug discovery have been extensively reviewed36, 37, 38 and are not discussed further here. More recently, collections or libraries of synthetic compounds have gained prominence in anticancer drug discovery efforts.
of action. In cancer treatment, natural products constitute several major therapeutic classes including the vinca alkaloids, camptothecins, and taxanes. Natural product extracts as sources for cancer drug discovery have been extensively reviewed36, 37, 38 and are not discussed further here. More recently, collections or libraries of synthetic compounds have gained prominence in anticancer drug discovery efforts.
Again, there are distinct advantages to each type of compound source. Synthetic compound libraries consist of easily identified single compounds and are usually amenable to assignment of activity to a unique chemical structure by a facile deconvolution algorithm. On the other hand, they are limited by the pharmacophore(s) around which the library has been constructed. Although natural product extracts have the possibility of remarkable diversity and stereochemically unique scaffolds, in some cases they require reacquisition from rather exotic ecological niches, optimization of fermentation or extraction approaches, and deconvolution of the active molecule(s) in the extract before the lead can be meaningfully pursued. The first step is the assignment of a structure for the active principle, often a tedious and challenging process. These strengths and limitations must be carefully considered when evaluating sources for screening endeavors.
The number and variety of synthetic compounds available for screening endeavors has been magnified by the use of combinational chemistry. The method of deliberately synthesizing more than one compound as a result of a single reaction began approximately 25 years ago with the synthesis by Geysen et al.39 of multiple peptides on polyethylene rods. These early endeavors, confined to the synthesis of small peptide libraries, were of limited value to cancer drug discovery because peptides tend not to enter cells and generally are not suitable drug candidates. Subsequent advances in the field included development of more efficient ways to track the compounds, use of a greater number of scaffolds on which to construct libraries, and use of a wider variety of reagents and amenable reaction conditions. Now, combinatorial chemistry provides an efficient method of exploring chemical space in a focused manner and, when applicable, an excellent means of rapidly defining structure activity relationships around active compounds. A somewhat newer trend is the use of “fragment libraries” where chemical fragments with potentially drug-like properties are screened at high concentrations to find scaffolds that are then chemically elaborated to increase potency.40,41
Advances in computer-based analysis of the physical properties and interactions of a novel, active compound have provided powerful tools for the pursuit of new anticancer drugs. Medicinal chemists traditionally have used parameters such as steric bulk, hydrogen-bonding ability, and hydrophobic interactions in the design of new drugs. The target pharmacophore now can be further refined by supplementing the information from these physical properties with biologic factors such as the crystal or solution structure of the target enzyme or receptor, the nature of the ligand, and the mechanism of the target-ligand (or target-inhibitor) interaction. Moreover, computer programs can create “virtual libraries,” which can be evaluated for exploring biochemical, functional space and the fit of proposed structure into that space. The use of computer models to design and filter novel structures can be a very efficient mechanism to increase the odds of identifying a potent and selective inhibitor of a well-defined target as recently exemplified by Keiser et al.42
Compound Libraries
Over the years, collections of pure compounds and natural product extracts coalesced into libraries, arrayed into 96-well plates or 384-well plates suitable for HTS. Popular commercial libraries are available from ChemBridge Corp. (San Diego, CA), Maybridge (Cornwall, England), and Sigma-Aldrich (St. Louis, MO; notably the Library of Pharmacologically Active Compounds or LOPAC), many of which may be cost prohibitive for academic researchers. Additional smaller collections of compounds are commercially available from an assortment of other suppliers. The NCI Open Source Repository (OSR) of approximately 250,000 samples is the publicly available subset of the compounds obtained by the NCI Developmental Therapeutics Program (DTP) over the past 50 years for use in anticancer drug screening (http://dtp.nci.nih.gov). Smaller subsets of the DTP collection, including diversity sets and collections of approved oncology agents, are also available; for both nonprofit and small business entities (costs are limited to shipping).
A major challenge for all suppliers of compounds is authentication of the structure and quantification of the purity of library materials. For the costlier commercial collections, one can expect suppliers to have undertaken appropriate quality control measures. Most compound sets derived from historical libraries of compounds show evidence of the toils of time, storage conditions, and questionable provenance. Most samples in the NCI compound repository were donated by academic chemists or industrial organizations, were accepted without further chemical characterization, and were stored at room temperature unless other conditions were specified by the supplier. As a result, the samples in this collection range in quality from those with a very high degree of sample integrity (purity and authenticity of structure) to those with little or none of the substance indicated by the supplier actually in the bottle. For such collections, verifying the structure and purity of selected “active” compounds may be easier than characterizing the entire library.
Numerous algorithms exist by which the diversity of a set of compound structures can be measured; however, little agreement is found regarding the best approach. In general, software programs partition libraries into a uniform array of blocks or cells on the basis of descriptor coordinates, and the number of cells is proportional to the level of diversity. Some algorithms define atom pair fingerprints, which indicate the presence or absence of pairs of atom types separated by a defined number of bonds, and use them to describe and differentiate each structure. Other algorithms cluster structures into groups; the Jarvis-Patrick clustering algorithm requires that each member of a cluster have in common a predefined number of chemical neighbors.43 The similar Hodes clustering model has been used by the NCI to assess structural novelty of new compounds submitted for screening.44 Often, the goal of these computer algorithms is to define a library of the smallest number of compounds that covers the greatest diversity of “molecular space.” Using these approaches, large collections of compounds can be winnowed into smaller sublibraries. However, detractors point out that by using these, for the most part, unproven tools and limiting the compounds screened, one risks decreasing the chance of finding drug leads. On the other hand, the economics of HTS calls for prudent use of reagents and encourages the use of such approaches.
A detailed analysis of the NCI OSR was conducted, with special emphasis on the uniqueness of the library relative to commercial libraries and other databases.45




Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
