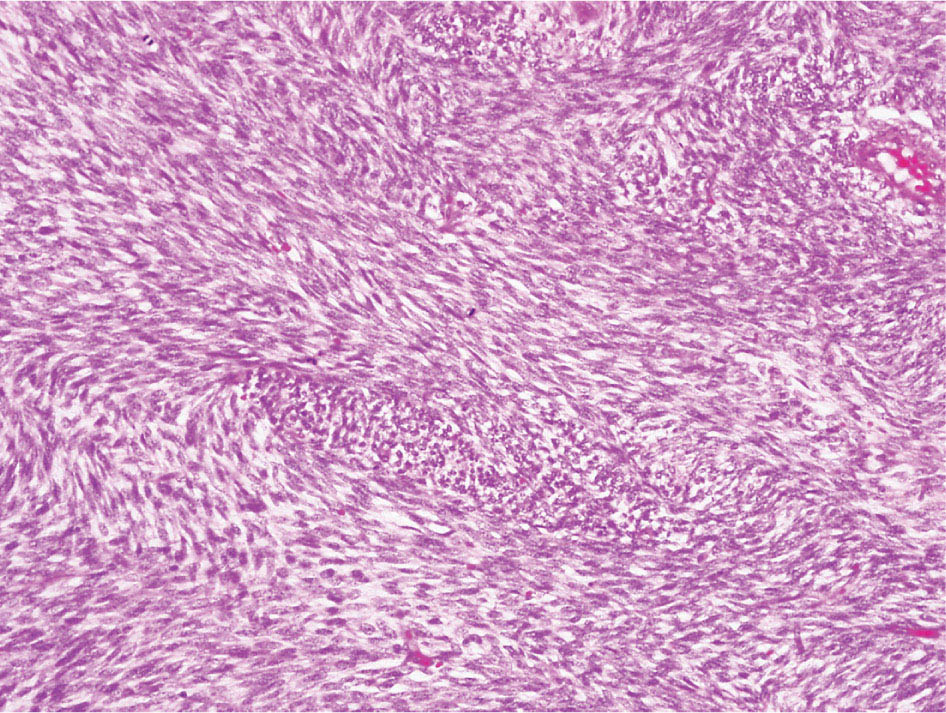
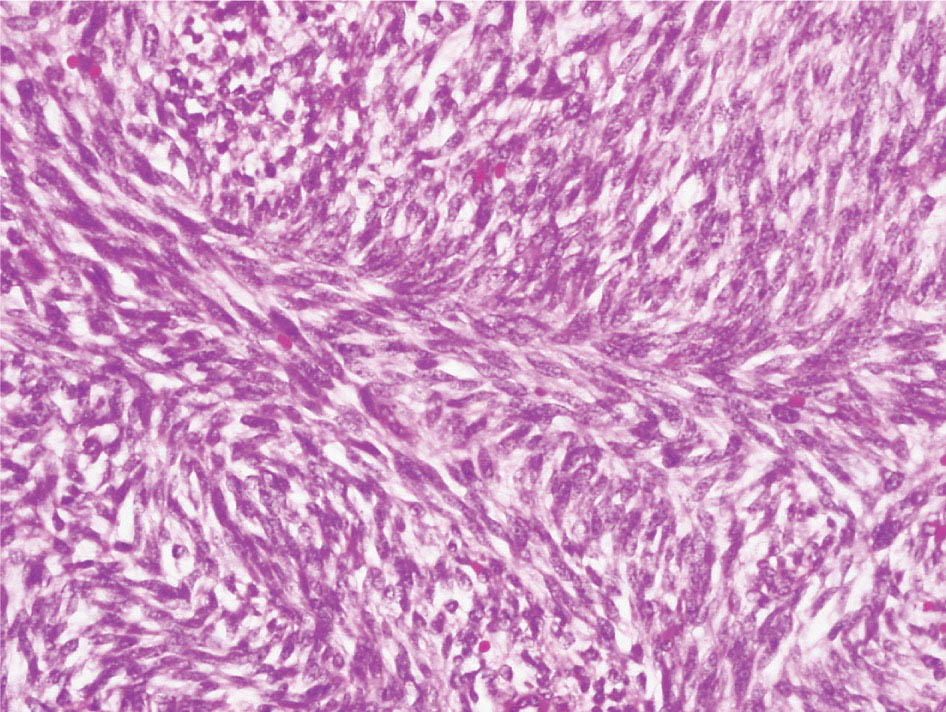
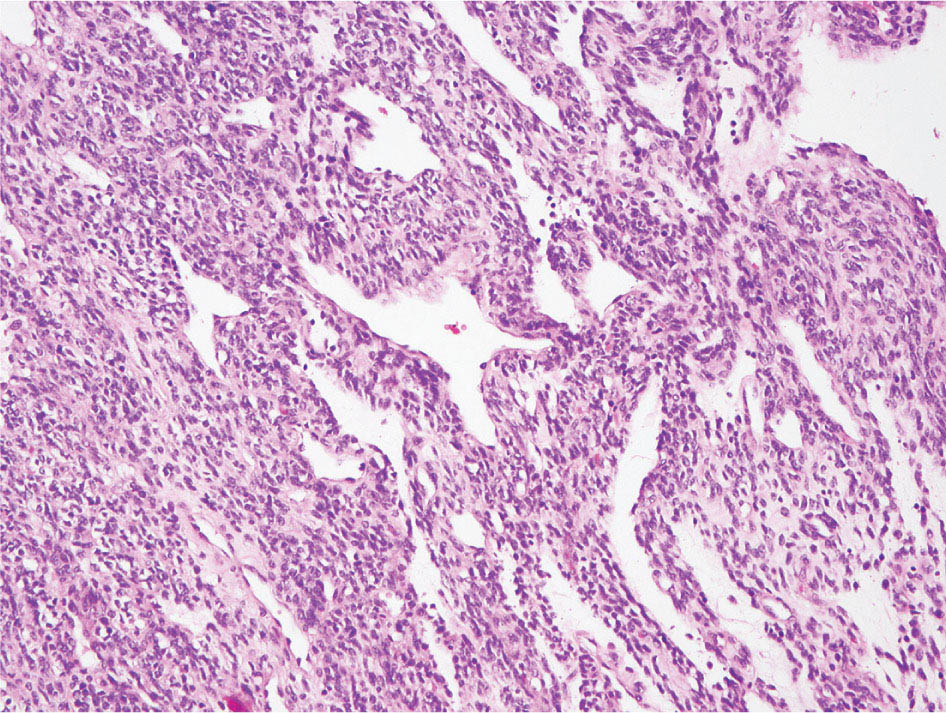
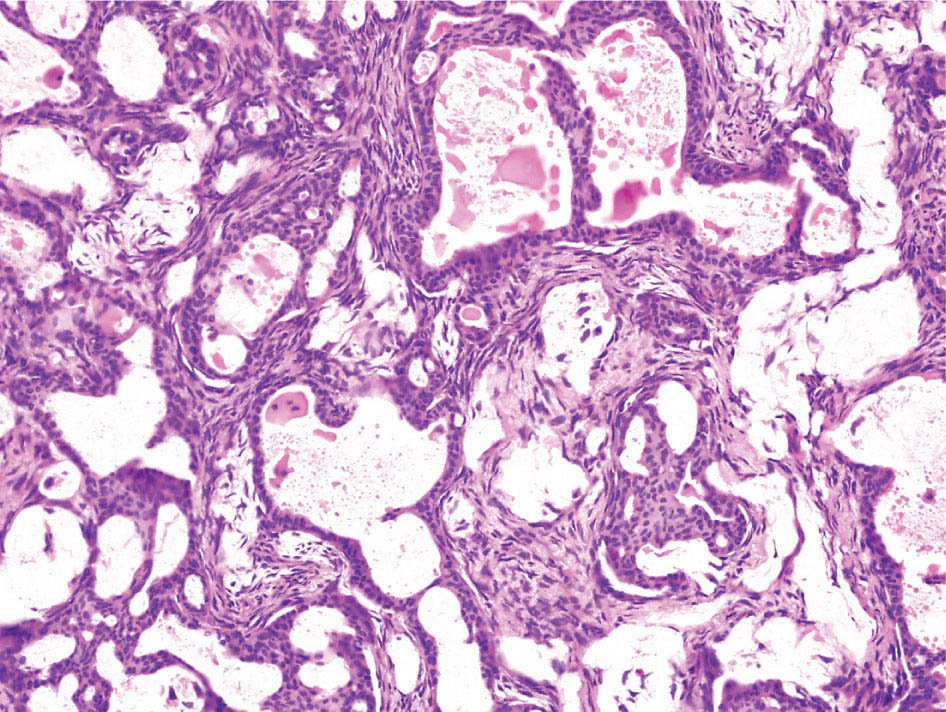
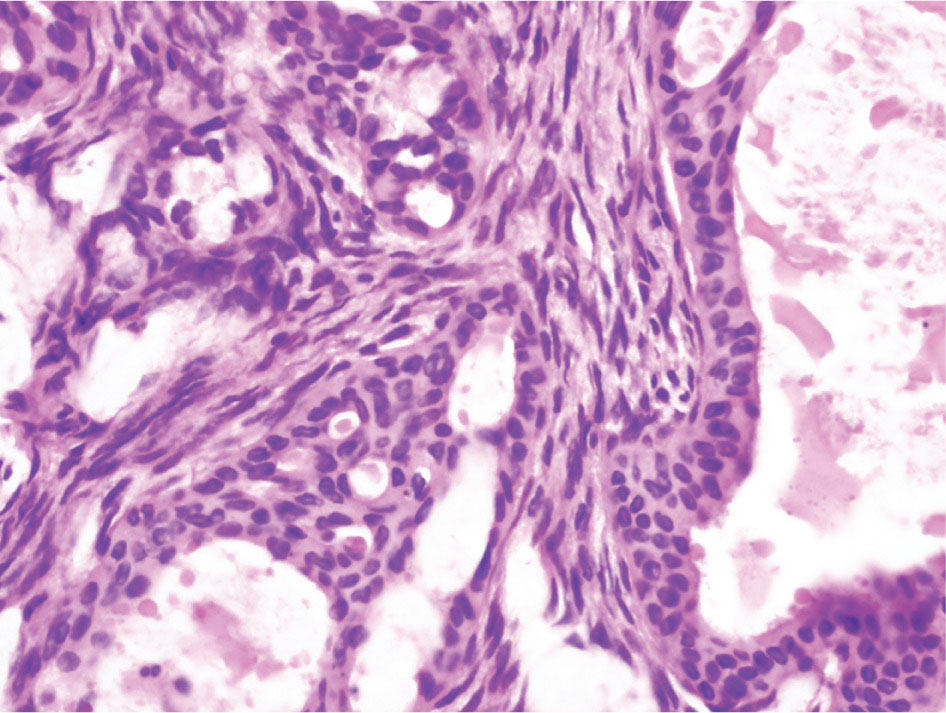
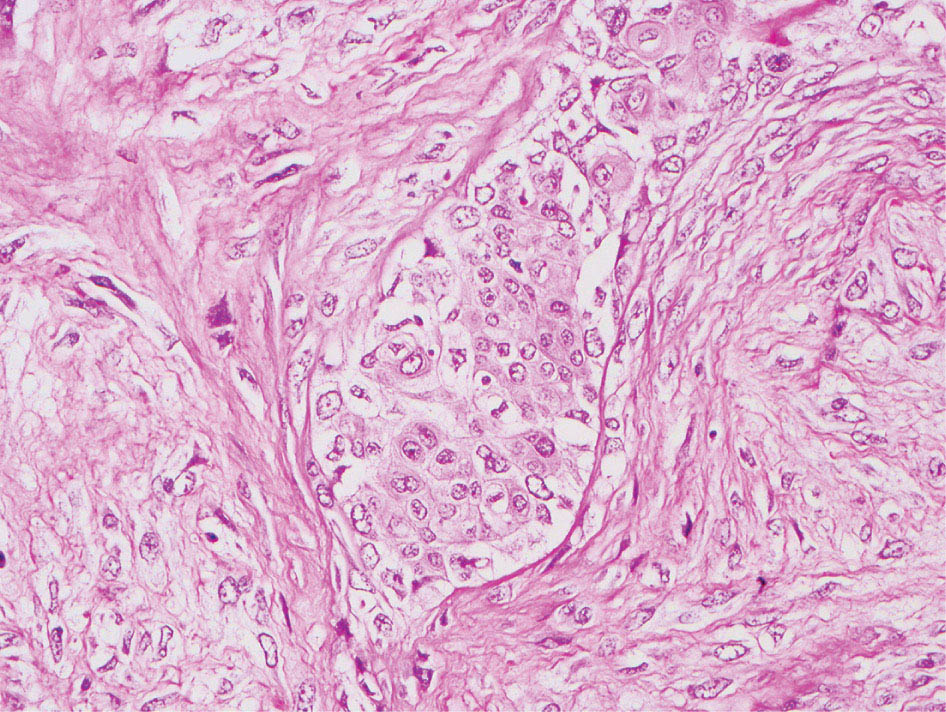
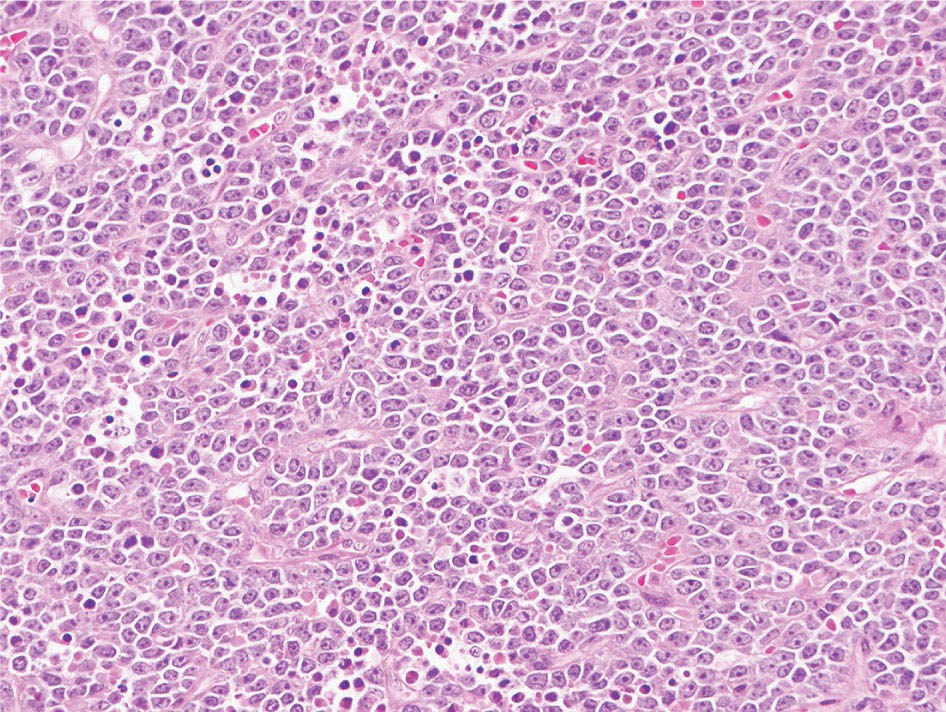
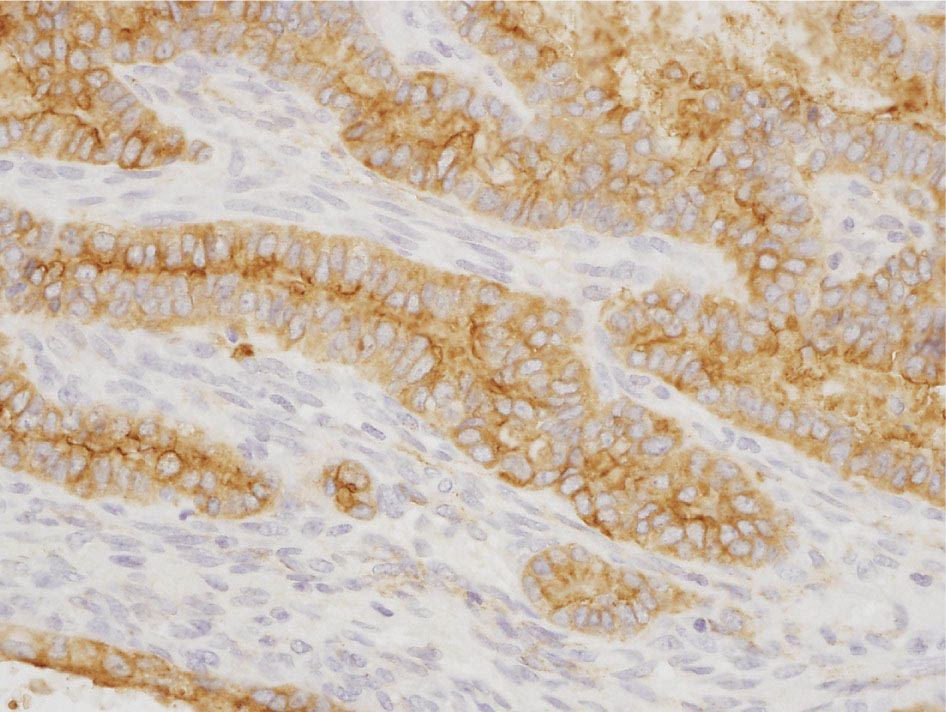
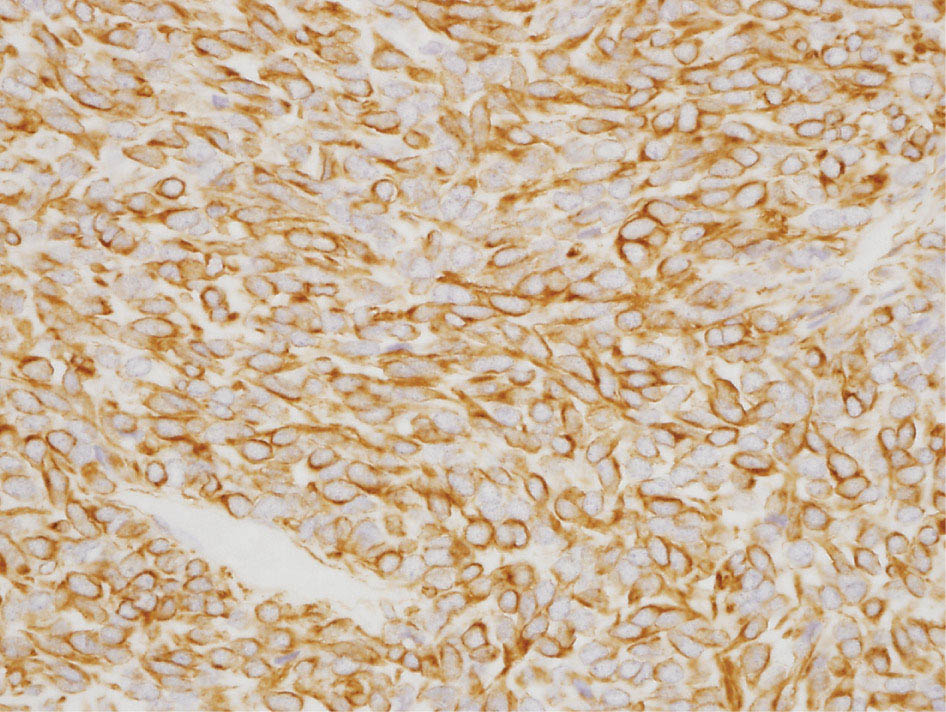
34327 Synovial Sarcoma Synovial sarcoma has unique features that distinguish it from other soft tissue sarcoma subtypes. It is diagnosed in both pediatric and adult patients. Under the microscope, it can manifest a variety of histologic features, ranging from epithelial cells to small, round cells. It is driven by a characteristic chromosomal translocation, which can serve as an important diagnostic feature. Correct diagnosis is important, and molecular diagnostic tools play an important role in making a diagnosis of synovial sarcoma. Synovial sarcoma is an aggressive disease, with propensity for metastatic spread. At the same time, it is considered a chemotherapy-sensitive type of soft tissue sarcoma. In the setting of high-risk, localized disease, multimodality therapy, including chemotherapy and radiation therapy, should be considered along with complete excision by an orthopedic oncologist or surgical oncologist with sarcoma expertise. Treatment options for metastatic soft tissue sarcoma, including synovial sarcoma, are increasing, including exciting immunotherapy advances. This chapter reviews diagnostic approach, pathologic features, molecular pathogenesis, and prognosis of synovial sarcoma, as well as therapeutic approaches for localized disease and metastatic disease. chemotherapy, chromosomal translocation, immunotherapy, patients, radiation therapy, synovial sarcoma Drug Therapy, Immunotherapy, Patients, Radiotherapy, Sarcoma, Synovial, Translocation, Genetic INTRODUCTION Synovial sarcoma has unique features that distinguish it from other soft tissue sarcoma subtypes. It is diagnosed in both pediatric and adult patients. Under the microscope, it can manifest a variety of histologic features, ranging from epithelial cells to small, round cells. It is driven by a characteristic chromosomal translocation, which can serve as an important diagnostic feature. There are various chemotherapy options for treatment of synovial sarcoma. In addition, it frequently expresses an antigen that has placed it at the leading edge of cellular immunotherapy research in sarcoma. EPIDEMIOLOGY Synovial sarcoma accounts for 5% to 10% of soft tissue sarcoma cases, with an incidence of approximately 0.13 per 100,000 person-years.1–3 Peak incidence of synovial sarcoma is in the third or fourth decade of life. It is the most common non-rhabdomyosarcoma soft tissue sarcoma among children and adolescents; one-third of synovial sarcoma cases occur in the adolescent-young adult (AYA) age group.1,4,5 Male and female incidence is similar.2,6 CLINICAL PRESENTATION Typically, synovial sarcoma arises in the deep soft tissues of the extremities. Less commonly, these tumors originate from the trunk, thorax, and head/neck.5–7 Like other sarcoma types, synovial sarcoma can arise almost anywhere in the body, with rare presentations, including, pleuropulmonary,8 cardiac,9 renal, gastrointestinal tract,10 prostate,11 and skeletal12,13 primary sites. Extremity synovial sarcoma typically presents as an enlarging mass. In a Surveillance, Epidemiology, and End Results (SEER) analysis of synovial sarcoma cases, 66% of patients presented with localized disease.6 Regional lymph node involvement is reported but uncommon.14 Despite the nomenclature, synovial sarcomas are not associated with the synovium and rarely involve the joint. DIAGNOSTIC APPROACH A pretreatment biopsy should be obtained to determine a tissue diagnosis and facilitate multidisciplinary treatment planning. This should be performed by an experienced surgeon or radiologist. The method of biopsy in synovial sarcoma depends on the location of primary disease. Historically, open biopsy was done to diagnose and grade sarcomas; however, in the extremity, core-needle biopsy is a reliable method for accurate diagnosis. Advantages of image-guided percutaneous biopsy include decreased cost, time, and complications compared to open biopsy.15–17 Excisional biopsy should not be performed, as the likelihood of positive margins is high, which may compromise management of the disease process.17 In addition, percutaneous biopsy for sarcoma minimizes the amount of tissue excised during resection of the biopsy tract at the time of surgical management.18 Like their extremity counterpart, retroperitoneal synovial sarcomas are diagnosed with image-guided core biopsy. However, if biopsy or imaging findings are inconclusive, resection may be performed to acquire a definitive diagnosis. 344RADIOGRAPHIC FEATURES Radiographically, synovial sarcomas may have calcifications located within the primary tumor, but typically do not have additional distinctive features from other subtypes. Imaging of the primary site with MRI or, if unable to obtain an MRI, CT scan is integral to determine disease extent and resectability of the tumor. MRI delineates not only tumor extent, but adjacent swelling as well. On MRI, synovial sarcomas are classically dark on T1-weighted sequences and bright on T2-weighted images. Although synovial sarcomas may occur in the retroperitoneum, the incidence is uncommon.19 In this anatomic location, both CT and MRI may be helpful to demarcate the entirety of disease and infiltration or impingement on adjacent structures.20 Chest imaging, with a chest CT without contrast (preferred) or chest x-ray study, should also be performed to evaluate for metastasis.21 When clinically warranted, PET/CT may be useful to characterize lung nodules and highlight other areas of disease not seen on CT.22 PATHOLOGIC FEATURES Origin Both neural and myogenic progenitor cells are candidate cells of origin for synovial sarcoma. Recent molecular studies correlating H3K4me3 levels and oncogenic gene activation in synovial sarcoma support a myogenic origin.23 Histology Synovial sarcomas appear as relatively nonspecific soft tissue masses, which present as relatively circumscribed tan-yellow, sometimes pink or bluish masses, which can have nonspecific areas of edema, hemorrhage, and calcifications.24 Synovial sarcoma has two main histologic appearances—biphasic and monophasic. Monophasic Synovial Sarcoma Monophasic synovial sarcoma accounts for approximately 70% of synovial sarcomas.25 Monophasic synovial sarcoma is a spindle cell sarcoma, composed of a very monotonous population of spindle cells devoid of any glandular elements (Figures 27.1 and 27.2). The spindle cells in synovial sarcoma usually grow forming fascicles and may adopt a wide variety of histologic appearances, including herringbone, storiform, hemangiopericytic, palisading, and sclerosing patterns. The tumor cells are characterized by the absence of nuclear pleomorphism or bizarre tumor cells. The spindle cells are usually small with carrot-shaped hyperchromatic nuclei. Mitotic activity may vary considerably in these tumors and can range from 1 to >10 per 10 high power fields; the majority, however, have low mitotic activity.24 Other features include stromal calcifications, abundant collagen, sclerotic changes, and myxoid degenerative changes. The appearance can be variable, and the diagnosis of monophasic synovial sarcoma needs to be eliminated in any tumor that resembles solitary fibrous tumor/hemangiopericytoma (SFT) because the cytologic features and the patterns of dilated, branching vessels can be similar in both entities (Figure 27.3). Likewise, some monophasic synovial sarcomas lack the SFT-like vascular patterns or have larger, ovoid nuclei and can resemble malignant peripheral nerve sheath tumor (MPNST) more than SFT.24 FIGURE 27.1 Monophasic synovial sarcoma shows fascicles of monotonous spindle cells with little intervening stroma. FIGURE 27.2 Higher magnification of monophasic synovial sarcoma showing uniform population of spindle cells displaying a herringbone growth pattern. FIGURE 27.3 “Hemangiopericytic” vascular pattern in a monophasic synovial sarcoma showing numerous dilated, sharp, angulated vessels that display branching. Biphasic Synovial Sarcoma Biphasic synovial sarcoma is characterized by an admixture of glandular structures with a compact, dense spindle cell proliferation in the stroma. A distinctive feature is that the glandular elements do not show a sharp segregation from the spindle cell elements, and the glandular structures are often flanked by strands of tightly packed atypical spindle cells. The glandular components can be quite subtle or very prominent. The presence of any glandular structures in these tumors qualifies for a diagnosis of biphasic synovial sarcoma (Figures 27.4 and 27.5). The glandular elements in biphasic synovial sarcoma can be of almost any type, including mucinous, tall columnar, simple cuboidal, with papillary excrescences, with cribriforming pattern, and with solid cords of tumor cells. Some cases can show squamous metaplasia (Figure 27.6) or areas of necrosis in the glandular component.24 Poorly Differentiated Synovial Sarcoma Poorly differentiated synovial sarcoma can arise from either monophasic or biphasic synovial sarcoma; however, a precursor lesion is not always present. It is characterized by round cell differentiation that can resemble those seen in lymphomas, Ewing sarcoma and embryonal rhabdomyosarcoma (Figure 27.7). Rhabdoid features, areas of necrosis, and a relatively higher rate of mitosis can be seen. The SFT-like vascular pattern is also often observed in poorly differentiated synovial sarcoma.26 FIGURE 27.4 Biphasic synovial sarcoma shows numerous irregular glands simulating a metastatic adenocarcinoma. FIGURE 27.5 Higher magnification of biphasic synovial sarcoma shows glandular structures surrounded by a monotonous spindle cell stromal proliferation, corresponding to the spindle cell component of the lesion. FIGURE 27.6 High-power magnification of biphasic synovial sarcoma in which the epithelial components show squamous differentiation with well-demarcated cell borders and intercellular bridges. FIGURE 27.7 Poorly differentiated synovial sarcoma simulating a small round blue cell tumor with sheets of round to polygonal atypical cells with prominent nucleoli. High mitotic activity can be seen in the background. Immunohistochemistry Immunohistochemistry has a limited role in the pathologic diagnosis of synovial sarcoma. Synovial sarcomas express both low- and high-molecular-weight cytokeratins (Figures 27.8 and 27.9). Cytokeratin expression is generally focal, and it can be confined to rare cells on the section. Immunostaining can be useful for revealing occult glandular differentiation in an otherwise monophasic appearing tumor. Epithelial membrane antigen (EMA) may be positive in some cytokeratin-negative monophasic and poorly differentiated synovial sarcomas.24 Other markers that stain synovial sarcoma include TLE1,27 BCL-2 (Figure 27.10), vimentin, epithelial membrane antigen, and calponin. Synovial sarcoma is generally negative for CD34. Molecular Features A nearly invariant feature of synovial sarcoma is the reciprocal translocation between the SS18 gene on chromosome 18 (18q11.2) and one of the three SSX genes (SSX1, SSX2, and rarely SSX4) that are clustered on chromosome X (Xp11.2).28,29 This translocation is unique to synovial sarcoma and is typically the only cytogenetic abnormality. It is detected in all morphologic subtypes and not observed in other human malignancies. This balanced translocation is believed to be the initiating molecular event leading to malignant transformation and development of synovial sarcoma. The translocation results in a fusion gene that codes for a chimeric protein composed of amino acids 1 to 379 of SS18 fused to the C-terminal 78 amino acids of SSX.30–32 Detection of this fusion event is pathognomonic for synovial sarcoma. The numerous reports in the literature correlating the specific SSX gene involved in the translocation and the histologic subtype of synovial sarcoma are inconsistent.33,34 FIGURE 27.8 Characteristic staining pattern of cytokeratin (AE1/AE3) in monophasic synovial sarcoma. The staining pattern is focal and patchy, with just a few scattered positive spindle cells. FIGURE 27.9 Immunohistochemical staining for cytokeratin (AE1/AE3) in biphasic synovial sarcoma shows strong positive staining of the glandular elements and a monotonous spindle cell proliferation in the background. Molecular Testing for Synovial Sarcoma Reverse transcriptase PCR has been demonstrated to be a highly effective and specific method for identifying the SS18–SSX fusion and is amenable to formalin-fixed, paraffin-embedded tissue.29,35 However, the success of reverse transcriptase PCR for the detection of the SS18–SSX translocation is highly dependent upon both knowledge of the target sequence of the primers and the quality of RNA isolated from the tumor. FIGURE 27.10 Strong and diffuse cytoplasmic positivity for BCL-2 in the majority of the tumor cells is also characteristic of monophasic synovial sarcoma. FIGURE 27.11 FISH assay demonstrating SYT translocation in synovial sarcoma using a dual color break-apart probe set with a 151-kb green probe and a 148-kb red probe flanking SYT at 18q11.2. Separate red and green signals indicate a chromosomal break at 18q11.2. FISH, fluorescence in situ hybridization. Fluorescence in situ hybridization (FISH)-based assays are an alternative approach to detect SS18–SSX translocations. These assays work well with archival or formalin-fixed, paraffin-embedded tissues in which the majority of tumor cells have interphase nuclei, and break-apart FISH probes are commonly used to detect disruption of the SS18 gene (Figure 27.11).36–38 In some instances, an atypical pattern consisting of loss of one of the probe signals is observed, and this should be interpreted as a peculiar unbalanced rearrangement of the SS18 gene, and subsequent SS18–SSX fusion testing should be considered.39,40 The highest analytical sensitivity and specificity (96% and 100%, respectively) have been reported by utilizing a combination of reverse transcriptase PCR and FISH.33 Immunohistochemistry for SYT and SSX1 proteins has been examined as an analytical method to distinguish synovial sarcoma from other soft tissue lesions. This approach is relatively sensitive— in one report, 41 of 47 cases of synovial sarcoma demonstrated strong nuclear expression of SS18.41 However, 19 of 99 non-synovial sarcoma cases included in the study also demonstrated variable nuclear and cytoplasmic staining with SS18, and these cases included malignant peripheral nerve sheath tumor, solitary fibrous tumor, and Ewing sarcoma, all of which are in the differential diagnosis of synovial sarcoma. Therefore, immunohistochemistry for the detection of synovial sarcoma has limited application for routine testing due to low specificity. Anchored multiplex PCR (AMP) is a novel technique for detecting gene fusion events using next-generation sequencing technology.42 This targeted RNA sequencing strategy utilizes AMP to detect oncogenic fusion transcripts without prior knowledge of fusion partners, making this approach an attractive option to reverse transcriptase PCR, in which precise knowledge of the coding sequence for each fusion partner is required. Furthermore, the assay supports multiplex analysis of multiple fusion events and detects both common and novel fusion genes.43 Concordance with FISH and reverse transcriptase PCR has been reported as high as 90% with advantages of this approach including superior performance and the identification of novel fusion partners.44 Differential Diagnosis The main differential diagnoses include MPNST, SFT, small blue round cell tumors, and fibrosarcoma. MPNST can so closely resemble monophasic synovial sarcoma that the two entities may be indistinguishable by light microscope. The absence of CD34 tends to support the diagnosis of monophasic synovial sarcoma, whereas a clinical history of neurofibromatosis favors MPNST. Classical examples of SFT will sometimes lack alternating hypocellular and hypercellular regions (“marbling” seen in MPNST), and express CD34. In equivocal cases, immunostaining for STAT6 will show positivity in SFT and will be negative in MPNST. In addition, MPNSTs frequently demonstrate loss of H3K27me3 while synovial sarcoma and solitary fibrous tumor retain expression making immunohistochemistry for H3K27me3 useful for differentiating MPNST from synovial sarcoma.45 Small blue round cell tumors can mimic poorly differentiated synovial sarcoma, but translocation analysis can now be used to distinguish synovial sarcoma from tumors such as Ewing sarcoma, desmoplastic small round blue cell tumor, and atypical Ewing sarcoma family tumors. Finally, fibrosarcoma, which is slowly disappearing 350as a diagnostic entity, is a diagnosis of exclusion to be reserved for cases in which histologic, molecular, and immunohistochemical analysis is uninformative. MOLECULAR PATHOGENESIS OF SYNOVIAL SARCOMA BAF Complex Chromatin regulation is a key process that controls the appropriate timing and levels of gene expression. This complex and highly ordered epigenetic process is achieved by modulating genomic architecture through histone modification, DNA methylation, and adenosine triphosphate (ATP)-dependent chromatin remodeling, thereby altering the accessibility of transcription factors to their cognate DNA binding sites. The BRG1/BRM associated factor (BAF) complex, which stands for Brg/Brm-associated complexes and is alternatively known as SWI/SNF complex, is one of the best characterized chromatin remodeling complexes. The BAF complex is a large, multiprotein, ATP-dependent chromatin remodeling complex and contributes to gene activation through nucleosome remodeling. The BAF complex assembles in a combinatorial manner involving 15 subunits coded by a set of gene families. Combinatorial assembly is tissue specific and plays an important role in determining cell fate.46–48 For example, using experimental techniques to force the formation of neural-specific BAF complexes converts fibroblasts to neurons.49 Mutations in the BAF complex are identified in 20% of all human cancers.50 Malignant rhabdoid tumors (MRTs) demonstrate biallelic inactivation of the BAF47 subunit, coded by the SMARCB1/INI1/SNF gene, in nearly all cases.50 BAF47 functions as a tumor suppressor gene and reintroduction of BAF47 into MRT cells stops cellular proliferation.51 Other examples include ARID1A52 and PBAF53 mutations, which are present in the majority of ovarian clear cell and renal clear cell carcinomas, respectively. The identification of specific BAF subunit mutations in specific malignancies suggests that the different combinatorial assembly of BAF complexes underlies the tissue-specific tumor phenotype.46 This is supported by the specialized role of BAF complexes in neural development and other cellular processes. SSX–SS18 Fusion Protein The SS18–SSX fusion protein effectively competes with the wild-type SS18 protein for incorporation and assembly into the BAF complex, and consequently, wild-type SS18 is displaced. Biochemical experiments suggest that SS18 and SS18–SSX do not compete for direct binding but rather the two proteins likely compete for assembly into complexes. In synovial sarcoma, the SS18–SSX outcompetes wild-type SS18 during assembly due to increased concentrations of the fusion protein.46 SS18–SSX-modified BAF also evicts the BAF47 tumor suppressor from BAF complex, where it is inactivated by proteasomal degradation, indicating that BAF47 is not required for proliferation of synovial sarcoma. SS18–SSX mediated disruption of the BAF complex is determined by two regions of the SSX protein: the C-terminal eight amino acids and two polar amino acids at positions 43 and 44. These two amino acids are conserved in SSX1, SSX2, and SSX4, but not in SSX3. In experimental models, SS18–SSX3 fusion proteins, which lack these two amino acids, can incorporate into BAF, but are deficient in eliciting BAF47 displacement, thus underscoring their role in BAF disruption.46 This is noteworthy from a therapeutic standpoint in that such a small region leads to complex disruption and may represent a potential target for the development of pharmaceutical inhibitors. The SS18–SSX-modified BAF complex demonstrates global genomic relocalization encompassing both the gain and loss of BAF target sites.23 The dual gain and loss of genomic sites of the SS18–SSX-modified BAF complex is a unique feature of synovial sarcoma. In contrast, BAF47-deficient tumors (for example, malignant rhabdoid tumors) demonstrate near exclusive loss of BAF complex occupancy on chromatin due to decreased BAF affinity.54 The key mechanism driving proliferation in synovial sarcoma is the gain of function activity and subsequent genome wide targeting of SS18–SSX-modified BAF complexes from enhancers, where wild-type BAF complexes normally localize, to polycomb repressive complex 2 (PRC2)-occupied domains resulting in the opposition of PRC2-mediated repression.23 This in turn leads to activation of bivalent genes, activation of the synovial sarcoma gene expression signature, and initiation of cellular pathways that drive proliferation. Furthermore, SS18–SSX-modified BAF opposition results in reduced PRC2-mediated methylation of H3 K27 and activation of bivalent genes including SOX2. SOX2, which is normally targeted for repression by PRC2, is universally expressed in synovial sarcoma secondary to SS18–SSX modified BAF and is essential for proliferation. 351The gene expression profile of synovial sarcoma is transcriptionally distinct from other cancers with BAF complex perturbations.23 Synovial sarcoma specific genes include TLE1, regulators of neural development, regulators of myogenesis (e.g., PAX3 and PAX7), and putative oncogenes (SOX2 and MYC). Transcription of these genes correlates with the respective proteins as determined by immunohistochemistry. Furthermore, MYC expression is mutually exclusive with PAX7 and SOX2. Expression of PAX7 and SOX2 is observed more frequently in primary synovial sarcoma, whereas MYC expression is more often observed in metastatic synovial sarcoma. Thus, SS18–SSX globally alters BAF-controlled gene regulation through a retargeting mechanism leading to inappropriate gene activation that drives synovial sarcoma proliferation and growth.55 Potential Therapeutic Implications Activation of bivalent genes by SS18–SSX-modified BAF is positively modulated by underlying H3K4me3 levels. This suggests reduction of H3K4me3 methylation via inhibition of MLL2/COMPASS as a potential therapeutic strategy to achieve abrogation of SS18–SSX-modified BAF-mediated gene activation.23 Furthermore, as BAF47 is evicted in SS18–SSX-modified BAF complexes and cellular levels are reduced to varying degrees, it is uncertain that selective inhibition of EZH2 (i.e., tazemetostat), which is indicated for BAF47-deficient solid tumors (e.g., malignant rhabdoid tumors), will demonstrate significant benefit for treatment of synovial sarcoma since its mechanism of sensitivity is based on INI1 depletion and EZH2 dependency. Synovial sarcoma is dependent upon a noncanonical BAF complex (ncBAF) for maintaining proliferation, implicating subunits of the ncBAF complex as a potential therapeutic target.56 Biochemical studies demonstrate that synovial sarcoma is dependent upon the ncBAF subunit BRD9, and synovial sarcoma proliferation is highly sensitive to perturbations of BRD9. This is significant in light of the development of selective BRD9 inhibitors.57–59 PROGNOSIS General Synovial sarcoma may behave aggressively, with reported 5-year survival rate of patients presenting with localized disease ranging from 60% to 70%.3,60,61 For patients with localized synovial sarcoma, larger tumor size, high grade, invasion of local structures such as bones or neurovascular structures, and axial primary tumor locations (as opposed to extremity tumors) are associated with increased likelihood of distant metastases and worse survival.62–64 The American Joint Committee on Cancer (AJCC) staging system incorporates factors such as tumor size and the presence or absence of regional lymph node or distant metastases to estimate prognosis. Lymph Node Involvement Historically, synovial sarcoma has been associated with elevated risk of regional lymph node metastases, with published rates greater than 13%,65 and has been grouped with other sarcoma subtypes, such as clear cell sarcoma, angiosarcoma, rhabdomyosarcoma, and epithelioid sarcoma as requiring particular attention to regional nodes. More recent analyses, including large data sets from SEER and the National Cancer Database (NCDB) as well as several large institutional databases, have found rates of regional lymph node involvement among synovial sarcoma patients ranging from 3.3% to 5.1%, similar to the overall rate for soft tissue sarcoma.66–70 Sentinel lymph node biopsy (SLNB) was evaluated in a prospective study of patients with localized soft tissue sarcoma subtypes with presumed high risk, but no clinical evidence, of lymph node involvement. Four of 68 patients with synovial sarcoma (5.9%) were identified as having lymph node metastases (three positive sentinel lymph nodes, one false negative). The investigators proposed that SLNB would not be relevant for patients with localized synovial sarcoma. Prognostic Tools Prognostic tools have been developed to help clinicians estimate sarcoma patient outcomes. A nomogram, developed at Memorial Sloan Kettering, uses patient and postoperative tumor characteristics (including histologic type) to estimate risk of sarcoma-related mortality.71 More recently, updated nomograms that estimate the risk of distant recurrence or death for patients with soft tissue sarcoma treated with curative intent have been developed and are the basis for Sarculator, a smart phone application that can be used as a prognostic tool.72 352THERAPEUTIC APPROACH: LOCALIZED DISEASE Surgery Surgery is the mainstay of treatment for all soft tissue sarcomas, including synovial sarcomas. Amputation is rarely necessary and limb-sparing resection is possible for the majority of patients.73 Resection should be performed by a fellowship-trained oncologic surgeon to ensure complete resection and minimize postoperative morbidity. Every attempt should be made to achieve a negative-margin resection to decrease the probability of local recurrence at the primary site. The goal of surgery should be an R0 resection with no ink on tumor. Acceptable local recurrence rates have been demonstrated in oncologically excised tumors with close margins of less than 1 mm.74,75 Careful dissection should take place through normal tissue planes without contaminating the resection bed by contacting the tumor. In the case of an open biopsy, the biopsy site should be resected. Nerves or vessels that are adjacent to synovial sarcomas do not need to be excised if the adventitia and perineurium are removed and there is no involvement of the underlying neurovascular structure. Surgical clips may be placed in the resection cavity to guide postoperative radiation, if warranted.17 Depending on the clinical scenario, radiation therapy may be performed in conjunction with surgery, either preoperatively or postoperatively, to decrease the risk of local recurrence. The utilization of radiation therapy should be strongly considered if close surgical margins are anticipated. In the clinical setting of metastatic disease, surgery may be considered based on performance status, disease burden, response to systemic therapy, and progression-free interval. Patients with oligometastatic disease or pulmonary-only disease may benefit from surgical management of symptomatic or bulky disease.76,77 Radiation Therapy External beam radiation therapy may be used in the preoperative or postoperative setting in synovial sarcomas. Although historically radiotherapy was delivered postoperatively, the several advantages of preoperative radiation include smaller field size, decreased dose and number of treatments, facilitation of surgical resection, and decreased fibrosis. One disadvantage, however, is the increased risk of postoperative wound complications.78 Radiation should be considered in tumors that are greater than 5 cm, high grade, or low-grade sarcomas that abut neurovascular structures. Additionally, radiation should be considered when a closer surgical margin will allow for a meaningful decrease in morbidity for the patient17 or when the risk of positive margins might result in tumor seeding, such as thoracic lesions, from which pleural seeding might occur. Brachytherapy delivers radiation through multiple catheters and may be considered; however, external beam is routinely used. Brachytherapy may be of particular benefit in the setting of local recurrence in a previously radiated field. Chemotherapy Although the use of chemotherapy in the adjuvant and neoadjuvant treatment of soft tissue sarcoma has been associated with both recurrence-free survival and overall survival, it has not been universally accepted by all sarcoma centers. Clinical trials have been difficult to interpret because of heterogeneous patient populations, variable treatment regimens, and small study sizes. A meta-analysis of data from studies of adjuvant anthracycline-based chemotherapy, published in 1997, found improved recurrence-free, but not overall, survival associated with chemotherapy.79 An updated meta-analysis reported statistically significant recurrence-free and overall survival benefit associated with adjuvant chemotherapy in the treatment of patients with localized, resectable soft tissue sarcoma. Benefit was most pronounced in patients treated with anthracycline and ifosfamide combination therapy, with reduction in risk of death with a hazard ratio of 0.56 (p = .01).80 Use of adjuvant chemotherapy has not been universally accepted because several older studies showed no significant benefit associated with adjuvant chemotherapy in the setting of localized, resected soft tissue sarcoma.81,82 Prospective, randomized trials that have shown perioperative anthracycline–ifosfamide chemotherapy to be beneficial share several features—patients have high risk soft tissue sarcoma (>5 cm, high-grade tumors), treatment is limited to histologic subtypes of sarcoma known to be chemotherapy responsive (including synovial sarcoma), and ifosfamide was given at adequate doses (for example ≥9 g/m2 per cycle).83,84 It is clear that there is a dose–response relationship with ifosfamide.85 The European Society for Medical Oncology (ESMO) sarcoma guidelines state that adjuvant chemotherapy be considered as an option with individual patients at high risk for poor outcomes.86 The 353National Comprehensive Cancer Network (NCCN) soft tissue sarcoma guidelines also suggest that neoadjuvant or adjuvant anthracycline-based chemotherapy be considered according to individual patient factors for stage II and III soft tissue sarcoma of the extremities and trunk.87 Additional data support the importance of anthracycline–ifosfamide chemotherapy, specifically in the treatment of localized synovial sarcoma. Several retrospective studies reported significantly better disease-specific survival for patients treated with ifosfamide-containing chemotherapy, compared to those who received no chemotherapy.88,89 Another study compared standard neoadjuvant epirubicin–ifosfamide chemotherapy to histotype-tailored chemotherapy, which was high-dose ifosfamide (14 g/m2 continuous infusion over 14 days, every 28 days) in the synovial sarcoma cohort. Standard therapy was associated with significantly better disease-free survival.84 FOLLOW-UP Follow-up for primary synovial sarcomas should involve a more rigorous schedule for several years after surgery, including interval history, physical exam, and baseline postoperative imaging of the primary site, with MRI or contrast-enhanced CT scan as an alternative with subsequent imaging based on the risk of recurrence and accessibility of the primary site to physical exam, and chest imaging every 3 to 6 months for the first 2 to 3 years, then every 6 months until year 5, and yearly thereafter. Owing to the propensity to develop late recurrence, follow-up is recommended for at least 10 years.17,90,91 The frequency of follow-up may be modified based on tumor location, patient function, tumor grade, and symptoms and clinical exam findings. METASTATIC RECURRENCE Synovial sarcoma has a propensity for metastatic spread. In several retrospective series, 40% to 45% of patients with localized synovial sarcoma have metastatic disease at 5 years.3,60,92 Median time to metastatic recurrence is less than 3 years, but metastases may also develop late, 10 years or more from initial diagnosis.3,60,92,93 Metastases are found in the lungs in at least 80% of cases, and much less commonly in regional lymph nodes and other sites, such as soft tissue, bone, and pleura.3,94 Metastatic synovial sarcoma is generally considered incurable, but there are some patients with limited extent of metastatic disease who may achieve long-term survival.93 THERAPEUTIC APPROACH: METASTATIC DISEASE Systemic therapy, with several cytotoxic chemotherapies or the multikinase inhibitor pazopanib, is commonly used in the treatment of metastatic synovial sarcoma. Radiation therapy, ablative therapies such as cryoablation, and surgery may be used in certain situations for disease or symptom control. Immunotherapeutic approaches are being studied and show significant promise. Chemotherapy Synovial sarcoma is considered a chemotherapy responsive sarcoma. In a review of first-line chemotherapy trials for advanced soft tissue sarcoma, it was noted that, compared to other sarcoma subtypes, synovial sarcoma patients experienced better objective response rates (28% vs. 19%), longer progression-free survival (PFS; 6.3 vs. 3.7 months), and longer overall survival (15 vs. 11.7 months).95 Anthracyclines, such as doxorubicin and epirubicin, are standard first-line treatment alone or in combination for advanced soft tissue sarcomas. Doxorubicin acts on cancer cells by inhibiting topoisomerase-II-mediated repair of DNA.96 As a single agent, at doses of 70 to 80 mg/m2 per cycle, doxorubicin achieves response rates of 14% to 20%.97–99 In a retrospective review of advanced synovial sarcoma patients treated at a sarcoma center, the response rate to single-agent doxorubicin was 25%.100 Olaratumab, a monoclonal antibody to platelet-derived growth factor alpha (PDGFRα), was granted Food and Drug Administration (FDA) approval in 2016, in combination with doxorubicin, for treatment of incurable soft tissue sarcoma. Approval was based on a randomized Phase II study that showed significant improvement in overall survival compared to single-agent doxorubicin (26.5 vs. 14.7 months).101 Unfortunately, the randomized Phase III study comparing doxorubicin to doxorubicin plus olaratumab found no benefit for the combination in response rates, PFS, and overall survival. 354Ifosfamide, an alkylating agent, at doses less than 10 g/m2 per cycle, is an active single agent treatment for advanced soft tissue sarcoma, with response rates ranging between 10% and 25% in several studies,85,102,103 and clear difference in response rates depending on dose, with doses of 5 g/m2 and 9 g/m2 per cycle defining the lower and upper ends of that range of response rates.85 For treatment of advanced sarcoma in general, it is used more frequently in combination with doxorubicin than as a single agent.104 Ifosfamide has particularly good antitumor activity in synovial sarcoma.95,105 In one retrospective review, among patients with advanced soft tissue sarcoma who had been previously treated with ifosfamide, there was treatment response noted on rechallenge with ifosfamide. Nearly half (14 of 29) of synovial sarcoma patients achieved partial response or stable disease.106 High-dose ifosfamide (HDI), 12 to 14 g/m2 per cycle, given as continuous infusion, has activity in advanced sarcoma as well, but is associated with significant toxicity including high rates of neutropenia fever.107,108 One study of HDI in first- or second-line treatment of advanced soft tissue sarcoma included 22 synovial sarcoma patients, eight of whom had objective responses.108 The combination of ifosfamide with anthracycline chemotherapy, for advanced soft tissue sarcoma in general, results in higher response rates and longer PFS, showed a trend toward an improvement in overall survival (1 year overall survival 60% vs. 51%, p = .07).99 Toxicity, including myelosuppression, is more pronounced with combination therapy. For these reasons, combination therapy is an option for synovial sarcoma patients with metastatic disease, particularly if a patient is symptomatic or has a large tumor burden. When doxorubicin/ifosfamide chemotherapy is assessed specifically in patients with synovial sarcoma, response rates are particularly impressive, with 88% of the synovial sarcoma subset of patients responding to combination therapy in one clinical trial,98 and 59% responding in another retrospective report.100 Pazopanib is an oral tyrosine kinase inhibitor with activity against vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF), which was FDA-approved in April 2012 for treatment of patients with metastatic nonadipocytic soft tissue sarcoma. Approval was based on the pazopanib for metastatic soft tissue sarcoma (PALETTE) trial, in which patients who had progressed on standard therapy were randomized to pazopanib 800 mg daily or placebo. Median PFS was significantly better in the pazopanib arm, 4.6 months versus 1.6 months.109 PFS in the synovial sarcoma subset was similar to that for the study population as a whole. While pazopanib is the only tyrosine kinase inhibitor FDA approved for treatment of metastatic soft tissue sarcoma, regorafenib, and sunitinib have demonstrated activity against synovial sarcoma as well.110,111 One example of this was a study of off-label use of targeted therapies by the French Sarcoma Group that included 15 patients with synovial sarcoma; eight of these patients were treated with sunitinib, and four had partial responses.112 Trabectedin was approved in 2007 by the European Medicines Agency and European Commission for treatment of advanced soft tissue sarcoma after failure of anthracycline and ifosfamide, and it was FDA approved in 2015 for advanced liposarcoma and leiomyosarcoma after failing anthracycline. FDA approval was based on a clinical trial that compared trabectedin to dacarbazine 1.5 mg/m2 as a 24-hour infusion every 3 weeks to dacarbazine. Median PFS was significantly longer in the trabectedin arm of the study, 4.2 months versus 1.5 months.113 A retrospective analysis of outcomes in patients with translocation-related sarcomas, treated in several Phase II trabectedin trials, included 45 synovial sarcoma patients. The synovial sarcoma patients had median PFS of 3 months.114 Multicenter retrospective review of synovial sarcoma patients treated with trabectedin included 61 patients, and found 15% objective response rate, median PFS 3 months, with 23% of patients without progression at 6 months.115 Gemcitabine-based therapy and various other cytotoxic chemotherapy agents are also used in the treatment of metastatic soft tissue sarcoma.116,117 There is insufficient data to report specific outcomes for synovial sarcoma patients. Tazemetostat, an EZH2 inhibitor, was studied in malignant rhabdoid tumors and sarcoma types with reduction or loss of INI1. There was limited activity in synovial sarcoma, with five of 33 patients achieving stable disease for 16 weeks or more.118 Immunotherapy Immune checkpoint inhibitors of PD-1 (programmed cell death protein 1) and CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) have been studied as treatment for soft tissue sarcoma patients. Two clinical trials, one with single-agent pembrolizumab and the other with nivolumab or a combination of nivolumab and ipilimumab, included 12 synovial sarcoma patients.119,120 One of these patients, treated with pembrolizumab, had brief tumor response.120 It is noted that, compared to undifferentiated pleomorphic sarcoma and dedifferentiated liposarcoma, synovial sarcoma expresses fewer inflammatory 355genes, has less PD-L1 (PD-1 ligand) staining, and lower T-cell fraction and T-cell clonality, suggesting synovial sarcoma is less likely to respond to these therapies. NY-ESO-1 is a cancer testis antigen expressed by various tumors, but with very limited expression in normal tissue, and it has been studied as a target for immunotherapy. Synovial sarcoma expresses NY-ESO-1 in 80% of cases.121 LV305 is a lentiviral vector NY-ESO-1 vaccine that targets dendritic cells, which then induce, or prime, an NY-ESO-1-specific T-cell response. CMB305, a prime-boost immunotherapy regimen, consisting of LV305 priming followed by repeated dosing with G305 (recombinant NY-ESO-1 protein combined with a TLR-4 agonist), has been studied in patients with NY-ESO-1 expressing solid tumors. This vaccine therapy was well tolerated, and 53% of the synovial sarcoma patients in the study had a best response of stable disease, with a 1-year survival rate of 86%.122 Another approach under investigation is genetic modification of autologous T cells so that the T-cell receptors recognize NY-ESO-1. This strategy is restricted to specific HLA type, HLA-A*02:01 or in another study HLA-A2*02:06.123,124 Objective responses among synovial sarcoma cohorts were noted in at least 50% of patients. Multidisciplinary Management of Metastatic Disease Pulmonary Metastectomy For patients with oligometastatic sarcoma, NCCN guidelines recommend consideration of metastectomy or use of other modalities, such as stereotactic body radiation therapy (SBRT) or ablation, in addition to management of the primary tumor and systemic therapy, as indicated.87 Lungs are a common site of sarcoma metastasis, and there are a number of reports of outcomes following pulmonary metastectomy. Median overall survival following pulmonary metastectomy ranges from 33 to 45 months,125,126 and 5-year survival rates range from 25% in a systematic review of the literature including all subtypes of soft tissue sarcoma to 58% in a single institution series of synovial sarcoma patients.127,128 Longer time from primary tumor treatment (>1 year), solitary metastasis, and complete resection of pulmonary metastases are factors associated with better survival after pulmonary metastectomy. Radiation Therapy SBRT may be an alternative when surgical excision of lung metastases is not feasible. Several series have been published, with varying radiation dosing regimens and follow-up periods, demonstrating lesion control rates of 82% to 96%.129–132 As there has not been randomized, prospective study of pulmonary metastectomy or SBRT as therapy for soft tissue sarcoma metastatic lesions, the true impact on survival is unclear. SUMMARY Synovial sarcoma can present in patients of any age, but it tends to occur in younger patients. Correct diagnosis is important, and molecular diagnostic tools play an important role in making a diagnosis of synovial sarcoma. Synovial sarcoma is an aggressive disease, with propensity for metastatic spread. At the same time, it is considered a chemotherapy-sensitive type of soft tissue sarcoma. In the setting of high-risk, localized disease, multimodality therapy, including chemotherapy and radiation therapy, should be considered along with complete excision by an orthopedic oncologist or surgical oncologist with sarcoma expertise. Treatment options for metastatic soft tissue sarcoma, including synovial sarcoma, are increasing, including exciting immunotherapy advances.


Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


