Abstract
Insulin resistance most commonly occurs in the context of obesity or other metabolic stressors, such as acute illness or inflammation; however, several known single gene defects are sufficient to produce severe insulin resistance in otherwise well, lean people. These mutations either affect genes directly involved in canonical insulin signaling ( INSR , PIK3R1 , AKT2 ) or indirectly impair insulin action by disrupting genes required for normal adipose tissue development and/or function (e.g., BSCL2 , AGPAT , PPARG , LMNA ). The characteristic syndromic features associated with the most severe impairment of insulin receptor function (Donohue syndrome and Rabson–Mendenhall syndrome) are readily recognizable; however, the more common, autosomal dominant form of insulin receptoropathy, which most commonly presents in peripubertal females as polycystic ovary syndrome associated with acanthosis nigricans and sometimes either hypoglycemia or diabetes, cannot easily be discriminated clinically from many other genetic subtypes of severe insulin resistance. Recently identified biochemical criteria, in particular normal or elevated serum adiponectin, normal blood lipid profile, and absence of fatty liver, now serve collectively to identify patients with INSR mutations with a high degree of accuracy, permitting targeted genetic testing. Autosomal recessive, congenital generalized lipodystrophies are also readily identified clinically; however, partial lipodystrophies are commonly underdiagnosed, particularly where central adiposity and head and neck adiposity is spared, and in lean men. Strong collateral clues to the presence of lipodystrophy may come from unusually severe hypertriglyceridemia or fatty liver, a history of recurrent acute pancreatitis, or reports of a “muscular” body appearance.
Keywords
insulin resistance, diabetes, lipodystrophy, insulin receptor, lamin, AGPAT2, BSCL2, PPAR, PCOS, hyperandrogenism
Introduction
Background, Incidence, Prevalence
Prospective studies have established that systemic insulin resistance (IR) is among the earliest detectable abnormalities in those who go on to develop type 2 diabetes, and consequently there is major interest in teasing out its genetics and molecular pathology. In the face of the burgeoning prevalence of obesity, IR is also increasing, and this may sometimes be severe. However, a small minority of patients have severe IR without obesity or any other obvious acquired precipitant. This is sometimes seen together with a failure of adipose tissue development, with abnormalities in adipose tissue distribution or function, or in the context of more complex recessive syndromes. For many of these conditions, a single genetic defect has now been defined. Primary forms of IR of known genetic basis are thus the focus of this chapter.
IR only produces diabetes in conjunction with beta cell decompensation, which may take decades to occur. Thus, although it is commonly brought to clinical attention as a failure to adequately control hyperglycemia despite large doses of insulin, the majority of patients are unrecognized in the prediabetic phase. Coupled to a common failure to appreciate the significance of the clinical signs of IR even once, hyperglycemia has supervened; this means that the syndrome is significantly underdiagnosed. This problem is compounded by the arbitrary biochemical thresholds often taken to denote severe IR. In some syndromes, characteristic clinical features permit diagnosis without reference to biochemical criteria, while in others the degree of hyperinsulinemia is critical. One set of operational diagnostic criteria is shown in Box 22.1 . Because of these complexities, no accurate prevalence figures for severe insulin resistance exist, though cumulative experience in one center suggests that it affects of the order of 0.5% of patients with type 2 diabetes.
- 1.
Nondiabetic and BMI <30 kg/m 2
Fasting insulin above 150 pmol/L or peak insulin on oral glucose tolerance testing above 1500 pmol/L.
- 2.
Absolute Insulin Deficiency and BMI <30 kg/m 2
Exogenous insulin requirement >3 U/kg/day.
- 3.
Partial Beta Cell Decompensation and/or BMI >30 kg/m 2
Insulin levels are difficult to interpret in the context of obesity, although comparison with sex and BMI-adjusted normal ranges is of use. Furthermore, in diabetes glucotoxicity, impairing islet function, and mixtures of endogenous and exogenous insulin in the circulation confuses the biochemical picture. In these settings the clinical history and features such as acanthosis nigricans assume particular importance in making a diagnosis of likely monogenic severe IR.
Clinical Presentation
Some clinical features of severe IR are generic to all known genetic forms of the condition, while some are particular only to some genetic subgroups. Although IR is normally thought of in terms of reduced ability of insulin to lower blood glucose, insulin is a highly pleiotropic hormone with a plethora of different metabolic and mitogenic effects, and this complexity is reflected in the range of its clinical manifestations. At least some of these appear to be due to preserved or enhanced signaling stimulated by very high insulin levels, either through the type 1 insulin-like growth factor (IGF) receptor, which has some ability to bind insulin, or via the insulin receptor through preserved arms of the intracellular insulin signaling network. The cardinal feature of severe IR is acanthosis nigricans, which is nearly a sine qua non for the condition, and is often associated with flexural skin tags (acrochordons). By far the commonest reason for seeking medical advice, however, is cosmetically distressing hirsutes and/or oligo- or amenorrhoea, driven by the effects of high levels of insulin, in tandem with gonadotropins, on the ovary. Indeed, ovarian hyperandrogenism may be severe, with serum testosterone concentration sometimes in the normal male range, and imaging usually reveals enlarged, polycystic ovaries.
A common clinical label used in cases of severe IR is “type A insulin resistance syndrome,” a term coined in the 1970s to denote lean women with the previously mentioned clinical features of severe IR, but without evidence of a causative circulating antibody indicative of “type B” extreme IR. “HAIR-AN” syndrome, another commonly used term, denotes “hyperandrogenism, IR and acanthosis nigricans,” and is thus essentially identical to the type A IR syndrome except that it has often come to be used only in women with BMI >30 kg/m 2 . This distinction is of some use, as there is a great enrichment of monogenic disease in lean, very insulin resistant patients; however, both descriptors in essence simply capture the general clinical features of severe IR described previously, and therefore, in isolation, do not imply any specific molecular defect.
Although monogenic severe IR most commonly presents as an aggressive form of polycystic ovary syndrome in young, lean women associated with acanthosis nigricans, diabetes is often diagnosed on oral glucose tolerance testing during the diagnostic work-up on the basis of hyperglycemia after the glucose challenge; however, not uncommonly fasting glucose may be low or normal. Indeed, a frequent but underrecognized early feature of severe IR is spontaneous and symptomatic postprandial hypoglycemia, which may be severe and require medical intervention, and which sometimes is reported to be exacerbated by metformin therapy. Only later in the natural history of severe IR does refractory hyperglycemia become the dominant problem, usually manifest as poor metabolic control despite very large doses of exogenous insulin. Because it often takes many years for beta cells to decompensate even in the face of severe IR, this may not occur until the fourth or fifth decade of life. Men are more likely to present at this stage than women.
The degree of IR in an individual with a monogenic defect is not invariant, and physiological or pathological influences that lead to IR often synergize with the inherited defect to exaggerate the clinical problem. Thus, puberty and the later stages of pregnancy, as well as intercurrent infection or illness, may in some cases lead to an increase in acanthosis nigricans and hyperandrogenism, and/or hyperglycemia, which is resistant even to huge doses of exogenous insulin.
The previous observations are true for all forms of severe IR. However, in some cases specific syndromic or biochemical features are present, which give a strong clue to the underlying single gene defect.
Primary Insulin Signaling Defects
Inherited defects in the insulin receptor most commonly present as type A IR, without obviously discriminating clinical features, although the lack of fatty liver or metabolic dyslipidemia, and preserved or elevated serum adiponectin concentration, are highly characteristic. In contrast, the most severe genetic defects in the insulin receptor present in early childhood or infancy. By virtue of their historical descriptions, which long antedated identification of the insulin receptor, the resulting constellations of clinical features are often classified either as Donohue syndrome (formerly leprechaunism; OMIM #246200) or Rabson–Mendenhall syndrome (OMIM#262190), though in truth there is a spectrum of clinical defects of which these two descriptions are arbitrary snapshots. In fact the clinical features of Donohue and Rabson–Mendenhall syndromes are remarkably similar, differing essentially only in the relative prominence of particular components of the syndromes. Features of the syndromes are summarized in Table 22.1 .
| Donohue | Rabson–Mendenhall | |
|---|---|---|
| Prognosis | Death in infancy | Death at 5–20 years |
| Metabolic abnormalities | Postprandial hyperglycemia; fasting hypoglycemia; extreme hyperinsulinemia; no ketoacidosis | Postprandial hyperglycemia; fasting hypoglycemia; later refractory hyperglycemia; extreme hyperinsulinemia; late ketoacidosis |
| Linear growth impairment | Low birth weight; postnatal growth retardation; severe failure to thrive | Low birth weight; postnatal growth retardation; short stature, low weight |
| Impaired development of tissues with high insulin-dependent glucose uptake | Paucity of adipose tissue; low muscle mass | Paucity of adipose tissue; low muscle mass |
| Soft tissue overgrowth | “Elfin” facies; large, low-set ears; prominent eyes; wide nostrils; thick lips; gingival hyperplasia; large mouth; acanthosis nigricans; large hands and feet; dysplastic nails; hypertrichosis | Coarse facies; prognathism; large, fissured tongue; gingival hyperplasia; dental dysplasia; premature eruption of teeth; acanthosis nigricans; dry, lichenified skin; onychauxis; hypertrichosis; pineal hypertrophy |
| Visceral abnormalities | Abdominal distention; nephromegaly; hepatomegaly; Cholestasis; hepatic fibrosis nephrocalcinosis; islet of Langerhans hyperplasia | Abdominal distention; nephromegaly; hepatomegaly; nephrocalcinosis; islet of Langerhans hyperplasia |
| Overgrowth of sex hormone-dependent tissues | Breast hyperplasia (female); prominent nipples; cystic ovaries; juvenile ovarian granulosa cell tumor; Leydig cell hyperplasia; large penis; large clitoris | Large penis; large clitoris; cystic ovaries |
| Miscellaneous | Frequent infections; decreased lymphatic tissue; delayed bone age | Frequent infections; motor developmental delay; precocious puberty |
The consequences of severe loss of insulin receptor function can be grouped into metabolic and growth defects. Metabolism is characterized by severe hyperinsulinemia (one to three orders of magnitude higher than the reference range) with initial preprandial hypoglycemia and postprandial hyperglycemia eventually degenerating into sustained hyperglycemia. Infants appear to be protected from ketoacidosis, although this becomes a major and refractory problem in older children. Leptin levels are usually low or undetectable, while adiponectin is paradoxically elevated after infancy. Growth defects include impaired linear growth and poor development of adipose and muscle tissue, which heavily rely on insulin-stimulated glucose uptake, contrasting with pseudoacromegaloid overgrowth of many other soft tissues, with additional features such as hypertrichosis. Particularly prominent is exaggerated growth of androgen-dependent tissues, which is a consequence of the ability of extreme hyperinsulinemia to synergize with gonadotropin actions on the gonads even in the absence of the insulin receptor.
Genetic defects affecting a more distal insulin signaling component have proved to be extremely rare. A single family with a loss of function mutation in the serine threonine kinase encoded by AKT2, a key effector of many of the metabolic actions of insulin, showed severe insulin resistance and hyperandrogenisim, but unlike receptor defects they also showed femorogluteal lipodystrophy, fatty liver, and dyslipidemia. More recently, mutations in the C-terminal of the p85α regulatory subunit of phosphatidylinositol-3-kinase, a signaling enzyme that functions downstream from the insulin receptor as well as many other tyrosine kinases, were found to underlie SHORT syndrome (short stature, hyperextensibility, ocular abnormalities, rieger’s anomaly, and teething delay). Insulin resistance, sometimes severe, was described in many of the patients with PIK3R1 mutations, and lipodystrophy has also been reported to be common; however, their penetrance and natural history have yet to be fully defined, and this condition will not be discussed further in this chapter.
Lipodystrophy
Lipodystrophic syndromes encompass a heterogeneous group of conditions characterized by partial or complete absence of adipose tissue. They may be genetic or acquired, and are further classified according to the anatomical distribution of the lipodystrophy. IR is a feature of most, but not all, of these disorders and may be severe. Where it complicates lipodystrophy it presents as acanthosis nigricans and dysglycemia in prepubertal children, and has the same clinical features as described for the type A IR syndrome in postpubertal patients. As with all forms of IR, the clinical expression is more pronounced in women and may be both more subtle and delayed in men. As nonalcoholic fatty liver and dyslipidemia are present in the vast majority (perhaps all) of patients with insulin resistance related to lipodystrophy, their presence in an insulin-resistant patient should prompt careful assessment of fat distribution and fat mass. Clinical and biochemical features of the most prevalent subtypes of congenital lipodystrophy are summarized in Table 22.2 .
| CGL | Familial partial lipodystrophy | |||
|---|---|---|---|---|
| Subtype | BSCL1 | BSCL2 | FPLD2 | FPLD3 |
| Defective gene | AGPAT2 | BSCL2 | LMNA | PPARG |
| Clinical onset | Soon after birth | Soon after birth | Puberty | Usually puberty, but may present in younger children |
| Fat distribution | Generalized absence | Generalized absence | Loss of limb and gluteal fat; typically excess facial and nuchal fat; trunk fat often lost | Loss of limb and gluteal fat; preserved facial and trunk fat |
| Cutaneous features | Acanthosis nigricans and skin tags; hirsutism common in women | Acanthosis nigricans and skin tags; hirsutism common in women | Acanthosis nigricans and skin tags; hirsutism common in women | Acanthosis nigricans and skin tags; hirsutism common in women |
| Musculoskeletal | Acromegaloid features common | Acromegaloid features common | Frequent muscle hypertrophy; some have overlap features of muscular dystrophy | Nil specific |
| NAFLD | Severe | Severe | Yes | Yes |
| Dyslipidemia | Severe associated with pancreatitis | Severe associated with pancreatitis | Yes, may be severe | Yes, may be severe |
| Insulin resistance | Severe early onset | Severe early onset | Severe | Severe; early onset in some |
| Diabetes onset | <20 years | <20 years | Variable; generally later in men than women | Variable; generally later in men than women |
| Hypertension | Common | Common | Common | Very common |
| Other | Mild mental retardation possible | |||
Congenital generalized lipodystrophy (CGL), also known as Berardinelli–Seip congenital lipodystrophy (BSCL), is characterized by a generalized absence of adipose tissue from birth. Children with the condition have increased appetite due to leptin deficiency, accelerated growth, and advanced bone age. Skeletal muscles, peripheral veins, and the thyroid gland are particularly prominent, due to the paucity of subcutaneous fat. Hyperinsulinemia is present from early childhood and leads to organomegaly and acromegaloid features as well as acanthosis nigricans. Diabetes tends to develop in the second decade. Hepatomegaly is often prominent and caused by severe nonalcoholic fatty liver disease (NAFLD), which generally progresses to nonalcoholic steatohepatitis (NASH) and even cirrhosis (believed to be the most common cause of death). In addition to the biochemical features of severe insulin resistance, these disorders are frequently characterized by severe hypertriglyceridemia, though this may be less striking in young children. In some cases, severe hypertriglyceridemia may be complicated by eruptive xanthomata and pancreatitis. This is a clinically useful way of distinguishing lipodystrophic syndromes from insulin receptoropathies as it essentially never occurs in the latter. Serum leptin and adiponectin concentrations are extremely low due to the lack of adipose tissue, although not, in themselves, diagnostic. The presence of myopathy (typically associated with an elevated creatine kinase) should lead to consideration of the possibility of a recently described subtype of CGL caused by mutations in PTRF. Although relatively few patients have been identified with this disorder, they also appear to exhibit less severe metabolic abnormalities than other forms of CGL.
Familial partial lipodystrophies (FPLD) have been classified into several subtypes of which the most prevalent include FPLD1 (Kobberling type; MIM #608600), FPLD2 (Dunnigan type; MIM #151660), and FPLD3 (MIM #603637). All three of these conditions are most readily detectable in postpubertal women where the loss of gluteal fat is particularly visually striking. They are very difficult to detect clinically in lean men. FPLD1 is characterized by loss of limb fat with preserved and frequently increased truncal fat. While some of these patients do have affected family members, many do not, suggesting that not all cases are inherited, and clinical observation suggests that additional factors such as the menopause and hyperandrogenism may be contributory. FPLD2 is a face-sparing lipodystrophy, which usually becomes apparent during puberty, although careful study of children known to harbor the underlying genetic defect in the LMNA gene suggests that fat distribution may also be subtly abnormal during childhood. The lipodystrophy predominantly affects the limbs and gluteal fat depots with variable truncal involvement but with normal or excess fat on the face and neck and in the labia majora. Metabolic abnormalities range from asymptomatic impaired glucose tolerance and mild dyslipidemia to severe insulin resistance with type 2 diabetes mellitus and severe dyslipidemia complicated by eruptive xanthomata and pancreatitis. As in the generalized forms of lipodystrophy, NAFLD/NASH is a common complication. Hypertension and accelerated atherosclerotic vascular disease have been reported in some kindreds.
FPLD3 is another disorder characterized by a paucity of limb and gluteal fat. It differs from FPLD2 in that abdominal fat is generally preserved and facial fat is often normal. Insulin resistance and lipodystrophy may be apparent in young children with this disorder, although the lipodystrophy more commonly only becomes clinically discernible during puberty in girls. Affected individuals are typically severely insulin resistant and manifest all the features of the metabolic syndrome including hypertension. Indeed, the very high prevalence of early onset hypertension discriminates FPLD3 from FPLD2 to some extent. NAFLD/NASH is almost universal and some patients manifest severe hypertriglyceridemia.
Recently, loss-of-function mutations have also been reported genes encoding lipid droplet coat proteins in even rarer patients with inherited partial lipodystrophies. The genes involved include CIDEC (only reported in one patient to date) and PLIN1 . The phenotypes of these patients were most similar to that of FPLD3.
Complex Syndromes
In addition to these conditions where severe insulin resistance and/or lipodystrophy are the dominant clinical manifestations of the underlying genetic defect, there exist a group of more complex syndromes that feature severe insulin resistance, often with some degree of lipodystrophy only as part of a wider spectrum of clinical abnormality. These include (1) mandibuloacral dysplasia, a rare disorder featuring short stature, mandibular and clavicular hypoplasia, dental abnormalities, acro-osteolysis, stiff joints, skin atrophy, alopecia, and mottled pigmentation as well as partial lipodystrophy, and some forms of progeria associated with biallelic LMNA or ZMSPTE24 mutations; (2) a somewhat similar syndrome characterized by subcutaneous lipodystrophy with sclerodermatous skin changes, sensorineural deafness, mandibular hypoplasia, and, in males, hypogonadism, due in all cases to de novo mutations in POLD1 , encoding the dominant lagging strand DNA polymerase; (3) Alström syndrome, a rare recessive disorder characterized by rod cone dystrophy, sensorineural hearing loss, heart failure, renal failure, and severely dyslipidemic insulin resistant diabetes, caused by mutations of a large centrosomal protein encoded by ALMS1; and (4) several syndromes of primordial dwarfism and/or defective DNA damage repair, including Bloom and Werner syndromes, osteodysplastic primordial dwarfism of Majewski type 2, and a recently described syndrome caused by mutation of NSMCE2. Each of these commonly feature severely dyslipidemic insulin resistance. Other extremely rare causes of syndromic severe insulin resistance/lipodystrophy have also been characterized, but these are beyond the scope of this chapter.
Genetic pathophysiology
Known INSR Mutations and Specific Phenotypes
The insulin receptor functions as a transmembrane dimer. The constituent monomers consist of disulfide-bonded alpha and beta subunits derived from the same allele, and the monomers in turn are also linked by disulfide bonds ( Fig. 22.1 ). The alpha subunit contains the insulin binding domain, while the intracellular beta subunit contains the tyrosine kinase domain, which autophosphorylates as a consequence of binding of insulin to the alpha subunit, thereby triggering a complex network of intracellular signaling events. The first pathogenic mutations in the insulin receptor were described in 1988, and since then over 150 different mutations have been discovered including missense, nonsense, and splice site mutations, insertions, and deletions.
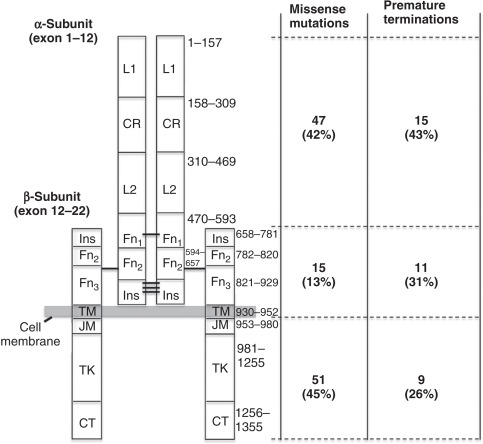
Collectively, published studies suggest that the clinical syndrome resulting from an insulin receptor mutation depends on the overall loss of receptor function, but beyond this there appears to be little specific genotype–phenotype relationship. However, there is no single cell-based assay that gives an entirely reliable global index of insulin receptor function, and consequently it is not possible to project clinical phenotype with complete confidence from commonly undertaken cellular studies. Furthermore, several reports have illustrated the potential of both genetic and environmental modifiers to influence the severity of the phenotype. Table 22.3 shows examples of mutations that have been reported as associated with Donohue, Rabson–Mendenhall, and type A IR syndromes, while Fig. 22.1 illustrates the approximate distribution of known naturally occurring mutations. It should be noted that the insulin receptor has a 27 amino acid signal peptide, which is cleaved during processing, and published reports variably number residues either according to the sequence of the mature receptor ( N ) or according to the transcriptional start site (i.e., N + 27). In this chapter, all numbering refers to the mature receptor. Further numbering confusion may arise in the literature because of the existence of two splice variants of the insulin, one containing 12 extra amino acids (first residue at position 745 in the mature receptor) in the alpha subunit due to the inclusion of exon 11, which is omitted in the other isoform.
| Syndrome | Mutations | Site |
|---|---|---|
| Donohue | Homozygous Entire INSR gene deletion: I29T, G31R, G84Q, R86P, R114W, K121X, Y134X, E124X, K148X, H209R, L233P, C259Y, S323L, del Val335, W412S, N431D, Q521X, R786X, L795P, V855X, R863X, T910M, R1092Q | 1 Large deletion; 17 α subunit; 6 β subunit (5 extracellular, 1 intracellular) |
| Heterozygous Q328X | 1 α-Subunit | |
| + 40 in compound heterozygous form | 10 Missense/missense; 10 nonsense/nonsense; 16 missense/nonsense; 4 other | |
| Rabson–Mendenhall | Homozygous R65W, R118C, I119M, P193L, C257Y, I321F, S323L, R735S, C807R, R1092Q | 8 α Subunit (including 1 at cleavage site); 2 β subunit (1 extracellular, 1 intracellular) |
| Heterozygous R1174W | 1 β Subunit (intracellular) | |
| + 22 in compound heterozygous form | 14 Missense/missense; 4 missense/nonsense; 4 other | |
| Type A IR IR-AN; HAIR-AN | Homozygous R118C, I119M, R252H, R252C, F382V, V1010L | 5 α Subunit; 1 β subunit (intracellular) |
| Heterozygous del exon 3, C225S, C253Y, R331X, S583W, S583N, Y675X, Y864X, del exon 14, del TK domain, del Leu999, G1008V, A1028V, A1028E, A1029G, A1048D, G1119R, A1121P, R1131W, R1131Q, A1134T, A1135E, A1137T, M1139K, G1149R, D1150H, M1153I, M1153K, R1155G, R1174Q, W1175R, P1178L, E1179D, R1182G, del T1187, W1193L, W1200S, P1209A, delT1214, Q1247X | 9 α Subunit; 31 β subunit (intracellular) | |
| + 28 in compound heterozygous form | 14 Missense/missense; 12 missense/nonsense; 2 other |
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


