Myelodysplastic Syndromes
Keywords
• Myelodysplastic syndromes • Cytogenetics • Molecular genetics • Diagnosis • Prognosis • Classification
The myelodysplastic syndromes (MDS) include a large spectrum of clonal hematopoietic stem cell disorders that are characterized by peripheral cytopenia(s), morphologic dysplasia, ineffective hematopoiesis, and a variable propensity to transform to acute myeloid leukemia (AML).1,2 The cytopenia can be limited to a single cell line, resulting in anemia, thrombocytopenia, or neutropenia, and can be chronic and somewhat indolent or profound, involving all 3 lineages with life-threatening consequences. The morphologic dysplasia associated with the ineffective hematopoiesis may be subtle and difficult to recognize, but, in some cases, it can be impressive and evident in both the bone marrow and peripheral blood. The variable increase in blasts relates, in part, to the risk for transformation to AML, although progression is not solely dependent on the blast percentage. MDS is a disease of older adults with a median age at diagnosis of approximately 70 years.3 Other risk factors for the development of MDS include tobacco use and exposure to solvents, such as benzene and agricultural chemicals. In approximately 10% to 15% of cases, the disease arises as a late complication of cytotoxic therapy (radiotherapy or chemotherapy) for a prior disorder, and is referred to as a therapy-related myeloid neoplasm. MDS is frequently associated with clonal cytogenetic abnormalities of significant prognostic4,5 and emerging therapeutic importance. An in-depth analysis of some of these is providing significant insights into underlying molecular alterations, which may hold clues to unraveling the pathogenesis of these disorders. This review focuses on the key role of cytogenetic analysis in MDS in the context of the diagnosis, prognosis, and molecular pathobiology of these disorders.
Diagnostic Considerations
MDS Classification
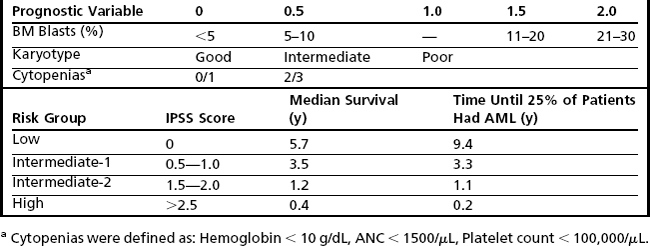
The earliest recognition of myelodysplastic disorders came with the identification of an anemia that was long standing and refractory, followed by the recognition that these were sometimes preleukemic.6 The classic myelodysplastic syndrome is exemplified in Fig. 1, which illustrates pancytopenia with multilineage dysplasia. Although such a case is classic, it belies the wide pathologic spectrum of MDS, which includes cases that are diagnostically challenging and difficult to distinguish, on one hand, from other benign causes of cytopenia and, on the other hand, from AML and other more aggressive clonal myeloid neoplasms.
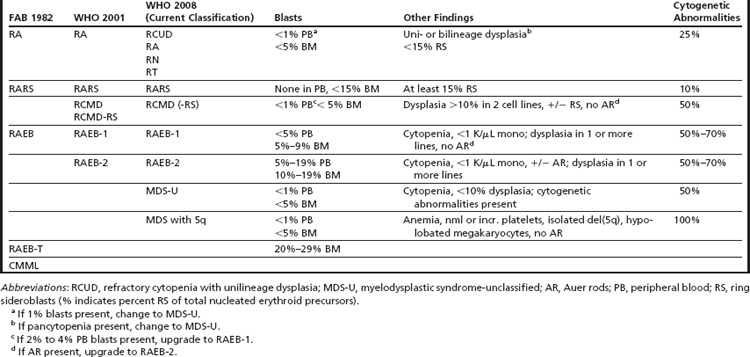
The disease spectrum was expanded with the first classification proposed by the French American British (FAB) group in 1982.7 In this classification scheme, the myelodysplastic disorders were divided into 4 subtypes with increasing blast percentage or as chronic myelomonocytic leukemia (CMML). The 4 entities included refractory anemia (RA), refractory anemia with ring sideroblasts (RARS), refractory anemia with excess of blasts (RAEB), and refractory anemia with excess of blasts in transformation (RAEB-T). These entities differed mainly by the percentage of blasts seen in the bone marrow (Table 1). In the FAB scheme, AML was defined by the presence of ≥30% blasts in the blood or marrow.
CMML was included by the FAB in the MDSs, although it was well recognized that CMML differed in that it had a proliferative component with increased circulating and marrow monocytes.8 At times, the peripheral leukocytosis was particularly elevated in the so-called myeloproliferative type, whereas the process was considered to be a dysplastic type and included in the MDS category when the count was less than 13 K/μL.9
The classification of MDS presented by the World Health Organization (WHO) committee in 2001 resulted in significant changes to the classification of both MDS and AML (see Table 1).10 Most notable was the reduction in the blast percentage required for a diagnosis of AML from 30% to 20% leading to the elimination of RAEB-T. The new classification also included a new subtype of MDS that, despite the lack of increased blasts (<5%), had a more aggressive course, probably owing to the presence of more pronounced multilineage dysplasia.11 This category was called refractory cytopenia with multilineage dysplasia (RCMD), and it comprised a substantial proportion of cases previously grouped in the low-grade RA and RARS categories. Although the recognition of RCMD as a relatively more aggressive MDS served to de-emphasize the importance of the blast percentage for prognosis, the new classification subdivided the RAEB category into 2 types, with 5% to 9% blasts (RAEB-1) and 10% to 19% blasts (RAEB-2), paradoxically emphasizing the prognostic significance of blast percentage in this category.12
A further significant change in the WHO 2001 classification scheme for MDS included the exclusion of CMML from the MDS category and the development of a separate nosologic group for CMML and other diseases in which there were features of both myelodysplasia and myeloproliferation.13 These “overlap” disorders are called myelodysplastic syndromes/myeloproliferative neoplasms and include CMML, “atypical CML,” and an unclassifiable category, which includes a provisional entity called RARS-T, a disorder that resembles RARS, but has associated thrombocytosis.
Further refinements in the WHO classification scheme for MDS were made most recently in 2008 (see Table 1).14,15 These included expanding low-grade MDSs from refractory anemia to refractory cytopenia with unilineage dysplasia, in which there is involvement of only 1 cell line. The 2008 WHO classification scheme also placed emphasis on the key role of cytogenetic analysis in the diagnosis of MDS, particularly in cases in which there is otherwise insufficient morphologic evidence to substantiate a diagnosis of MDS. This is reflected in the inclusion of the subtype, myelodysplastic syndrome unclassified, defined by the presence of cytopenia, less than 1% peripheral blasts, less than 10% dysplasia, and less than 5% bone marrow blasts with the presence of a cytogenetic abnormality commonly seen in MDS (Table 2). In addition, the WHO 2008 classification now includes “MDS with an isolated del(5q)” as a separate entity. This entity, sometimes referred to as the 5q− syndrome16 had been well known for some time and is characterized typically by its presentation in middle-aged women with macrocytic anemia, splenomegaly, normal-to-elevated platelet counts, hypolobated megakaryocytes in the bone marrow, and an isolated del(5q).
Prognosis
The heterogeneity in outcome within the various morphologic categories identified by the FAB and WHO classification systems has led to a proliferation of prognostic tools and scoring systems that attempt to predict outcomes of patients with MDS more accurately. The ability to achieve greater precision in prognostication in MDS is of paramount importance, because therapeutic options vary from supportive care and growth factor use to more intensive approaches, such as epigenetic modulators, and to approaches associated with significant potential for morbidity and mortality, such as intensive chemotherapeutic strategies and allogeneic stem cell transplantation. Currently, the higher-intensity approaches are reserved for patients with high-risk disease.17 All of the contemporary prognostic systems incorporate the cytogenetic pattern as a key element.18–21
The most widely used, and validated, prognostic scoring system in current use is the International Prognostic Scoring System (IPSS), which incorporates karyotype, bone marrow blasts, and number of cytopenias and identifies these factors as being the most critical in prognostication (Table 3).18 Nonetheless, there are a number of limitations that have led to the development of other scoring systems as well as ongoing attempts to refine the IPSS. Limitations of the IPSS include the fact that it was validated in previously untreated patients with de novo MDS, which limits the ability to use this tool to predict outcomes in patients with MDS treated using contemporary approaches. In addition, although the numbers of cytopenias are factored into the IPSS, the severity of cytopenias is not taken into account; in particular, transfusion dependency, which has been identified as a poor prognostic marker in recent scoring systems, is not considered. Furthermore, the IPSS includes a limited number of cytogenetic abnormalities (Table 4) and there are concerns that high-risk cytogenetic aberrations are not accorded sufficient emphasis.
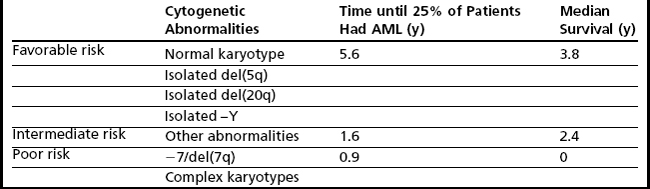
To address some of the concerns associated with the IPSS, newer systems, such as the WHO classification–based prognostic scoring system (WPSS) have emerged.20 The WPSS is dynamic, can be applied at diagnosis and during follow-up, and is now being validated in patients with MDS undergoing contemporary treatment modalities, such as allogeneic stem cell transplantation.22 Prognostic factors of import included in this model are WHO subtype, transfusion dependency, and karyotype subgroup, as defined by the IPSS. Five risk groups are recognized in the WPSS, with median survival times ranging from 12 months to 103 months.
The MD Anderson Cancer Center prognostic scoring system was also proposed recently to address some of the limitations associated with the IPSS.19 This model is also dynamic, can be applied at any time during the disease course, and encompasses a broader patient population, including patients with therapy related MDS (t-MDS) and CMML. Four risk categories are identified that predict overall survival, ranging from a median of 54 months in low-risk patients to 6 months in high-risk patients.
Despite the development of newer systems, there continues to be significant heterogeneity in outcome, particularly in patients identified by current models as being in the lower-risk category, emphasizing the need for continued refinement of these models based on a broader range of anomalies of emerging prognostic significance.23 An updated cytogenetic system has been proposed that involves an international collaborative effort using data sets originating from the German-Austrian Working Group, the International Risk Analysis Workshop, the Spanish Cytogenetics Workshop, and the International Cytogenetics Working Group of the MDS Foundation. This 4-tiered cytogenetic risk stratification system (Table 5) involves 20 cytogenetic subgroups; median survival ranged from 5.7 months to 50.6 months, and time until 25% of patients evolved to AML ranged from 3.4 months to 71.9 months.23
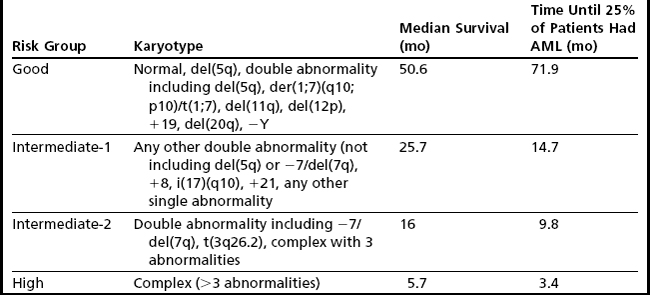
There is also ongoing concern that the IPSS accords more prognostic weight to an increase in bone marrow blasts compared with poor-risk karyotypes, for example, 2 points are assigned for bone marrow (BM) blasts in the 21% to 30% range, and 1.5 points for the 11% to 20% range, whereas poor-risk karyotypes are assigned 1 point (see Table 3). In a recent retrospective study of 2351 patients with MDS, the prognostic impact of a poor-risk karyotype was as unfavorable as the presence of a significant increase in blasts greater than 20% blasts.24
In addition to the broad prognostic variables incorporated in the various systems outlined above, it is clear that gene mutations25 and epigenetic aberrations play a critical role in the pathobiology of MDS 26,27 including the determination of prognosis. These genetic and epigenetic determinants, described in more detail in the following sections, deserve consideration for inclusion in a contemporary prognostic model developed for 21st century use.
Cytogenetic Analysis
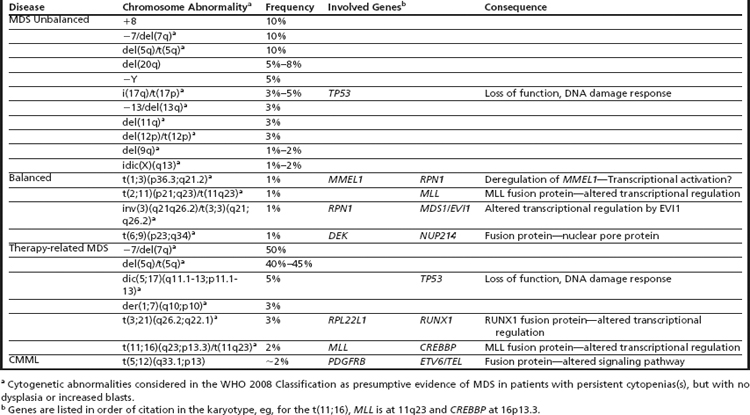
Clonal chromosome abnormalities can be detected in marrow cells of 40% to 100% of patients with primary MDS at diagnosis (see Tables 1 and 2).5,18 The proportion varies with the risk that a subtype will transform to AML, which is highest for RCMD and RAEB. Most recurring cytogenetic abnormalities found in MDS are unbalanced, most commonly the result of the loss of a whole chromosome, a deletion of part of a chromosome, or an unbalanced translocations (Fig. 2). The most common cytogenetic abnormalities encountered in MDS, del(5q), −7, +8, and del(20q), have been incorporated into the more robust prognostic scoring systems of MDS outlined above. The recurring translocations characteristic of acute leukemia without prior MDS, such as the t(15;17), inv(16), and t(8;21), are almost never seen. With the exception of MDS with isolated del(5q), the chromosome changes show no close association with the morphologic subtypes of MDS. Clones with unrelated abnormalities, one of which typically has +8, are seen at a greater frequency in patients with MDS (5% vs 1%), than in patients with AML.
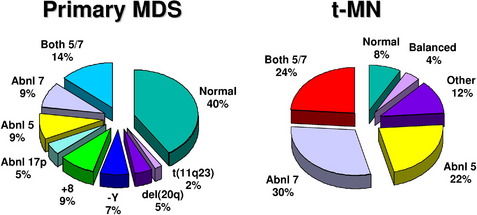
Fig. 2 Recurring chromosomal abnormalities in MDS. Relative frequencies are depicted in the pie chart.
Serial cytogenetic evaluations can be informative, particularly when there is a change in the clinical features of a patient. Cytogenetic evolution is the appearance of an abnormal clone in which only normal cells have been seen previously, the acquisition of additional abnormalities within an abnormal clone, or the progression from the presence of a single clone to multiple related, or occasionally unrelated, abnormal clones. Karyotypic evolution in MDS is associated with transformation to acute leukemia in about 60% of cases and reduced survival, particularly for those patients who evolve within a short period of time (<100 days).5,18
Fluorescence In Situ Hybridization Analysis
In the last decade, molecular cytogenetic techniques, such as fluorescence in situ hybridization (FISH) have become a powerful adjunct to classical cytogenetic analysis.28 FISH can be performed on marrow or blood smears or fixed and sectioned tissue because it does not require dividing cells. Advantages of FISH include (1) the rapid nature of the method and the ability to analyze large numbers of cells, (2) its high sensitivity and specificity, (3) the ability to obtain cytogenetic data from samples with a low mitotic index or terminally differentiated cells, and (4) the application to histologically stained cells allowing a direct correlation of the status of the genetic target within morphologically characterized cells. The major disadvantage is the inability to interrogate more than a few abnormalities and to identify the full spectrum of abnormalities in each clone, the presence of multiple clones, and clonal evolution.28 In a clinical setting, cytogenetic analysis could be performed at the time of diagnosis to identify the chromosomal abnormalities in bone marrow cells from an individual with MDS. Thereafter, FISH with the appropriate probes could be used to detect residual disease or early relapse and to assess the efficacy of therapeutic regimens. Commercially available probes have been developed for the detection of 11q23/MLL translocations, −Y, del(5q)/t(5q), −7/del(7q), +8, del(20q), del(13q), del(11q), and −17/loss of 17p. In MDS, FISH analysis of cases with a normal karyotype by cytogenetic analysis detected recurring abnormalities in 10% to 15% of cases.29
Cytogenetic Findings in MDS
Normal Karyotype
A normal karyotype is found in approximately 50% of patients with MDS. This group of patients is almost certainly genetically heterogeneous, where technical factors precluded the detection of chromosomally abnormal cells, or where leukemogenic alterations occur at the molecular level and are not detectable with standard cytogenetic methods. Nonetheless, these cases are a standard reference for comparison of outcomes. The IPSS found that patients with a normal karyotype fall within the favorable risk group (see Table 4).18
−Y
The clinical and biological significance of the loss of the Y chromosome, −Y, is unknown. Loss of the Y chromosome has been observed in a number of malignant diseases but is also associated with aging in healthy men.30 Patients with a hematologic disease have a significantly higher percentage of cells with a –Y (52% vs 37%, P=.036), and –Y in greater than 75% of metaphase cells accurately predicted a malignant disease.30 Although loss of a Y chromosome may not be diagnostic of MDS, once the disease is identified by clinical and pathologic means, the IPSS found that −Y as the sole cytogenetic abnormality conferred a favorable outcome.18
+8
A gain of chromosome 8 in MDS is observed in all MDS subgroups in approximately 10% of patients.18,31,32 Determining the significance of the gain of chromosome 8 in MDS patients is complicated in that +8 is often associated with other recurring abnormalities known to have prognostic significance, for example, del(5q)t(5q) or –7/del(7q), and may be seen in isolation as a separate clone unrelated to the primary clone in up to 5% of cases. The IPSS,18 as well as the time-dependent WPSS20 ranked this abnormality in the intermediate-risk group; however, several subgroups found that +8 as a sole abnormality had a poorer outcome than expected for an intermediate IPSS risk group.31,33 Hematopoietic cells with trisomy 8 express higher levels of many genes that localize to chromosome 8, including the MYC gene, and antiapoptotic genes.
del(20q)
A deletion of the long arm of chromosome 20, del(20q), is seen in approximately 5% of MDS cases and 7% of t-MDS cases.5,31 Features characterizing MDS patients with a del(20q) include low-risk disease (usually RA), low rate of progression to AML, prolonged survival (median of 45 months vs 28 months for other MDS patients), and prominent dysplasia in the erythroid and megakaryocytic lineages.34 The IPSS noted that patients with a del(20q) in the context of a complex karyotype identified a poor-risk group (median survival, 9.6 months), whereas the prognosis for patients with an isolated del(20q) was favorable.18 Although a commonly deleted segment (CDS) has been identified on 20q, containing 19 genes, there is no definitive evidence linking these genes to the pathogenesis of myeloid neoplasms.35,36
Deletions of Chromosome 5
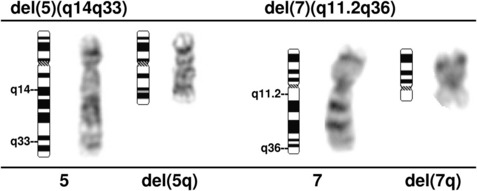
A deletion of the long arm of chromosome 5, del(5q), or an unbalanced translocation leading to loss of 5q are observed in 10% to 20% of MDS patients, more commonly in RAEB-1 and -2 in association with a complex karyotype, and 40% of patients with t-MDS (Fig. 3).37 Abnormalities of 5q are associated with a significant occupational exposure to potential carcinogens as well as previous exposure to standard and high-dose alkylating agent therapy, including use in immunosuppressive regimens.38,39 Clinically, the patients with del(5q) coupled with other cytogenetic abnormalities have a high frequency of TP53 mutations, a poor prognosis with early progression to leukemia, resistance to treatment, and short survival.40,41 MDS with an isolated del(5q) represents a distinct clinical syndrome, “the 5q- syndrome.” These patients have a favorable outcome, in fact, the best of any MDS subgroup, with low rates of transformation to AML and a relatively long survival of several years’ duration.16,18 The immunomodulatory agent, lenalidomide, has been associated with significant responses in IPSS low or intermediate-1 risk MDS with the del(5q), including red cell transfusion independency (67% of patients), and complete cytogenetic responses.42
Related posts:
 Cytogenetics
Cytogenetics
 Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
 Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
 In Situ Hybridization
In Situ Hybridization
 Myeloma: Current Perspectives
Myeloma: Current Perspectives
 Myeloid Leukemia: Conventional Cytogenetics, FISH, and Moleculocentric Methodologies
Myeloid Leukemia: Conventional Cytogenetics, FISH, and Moleculocentric Methodologies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


