Stephen H. Zinner, Kenneth H. Mayer Keywords brucellosis; drug interactions; human immunodeficiency virus/acquired immune deficiency syndrome (HIV/AIDS); malaria; methicillin-resistant Staphylococcus aureus (MRSA); Pneumocystis jirovecii; prophylaxis; Staphylococcus; Stevens-Johnson syndrome; sulfa allergy; sulfamethoxazole; sulfonamides; toxoplasmosis; trimethoprim; urinary tract infections
Sulfonamides and Trimethoprim
The modern era of antimicrobial chemotherapy began in 1932 with the first reports by Domagk of the protective activity of sulfachrysoidine (Prontosil) against murine streptococcal infections. This drug was developed initially by the German dye industry and had been available commercially since the early 20th century. Sulfachrysoidine exerted its antibacterial activity through the release in vivo of para-aminobenzenesulfonamide (sulfanilamide). This was the first antibacterial agent used in the United States, in July 1935, in an unsuccessful attempt to treat a 10-year-old girl late in the course of meningitis and sepsis caused by Haemophilus influenzae.1 During the late 1930s, the basic sulfanilamide compound was modified to remove unpleasant side effects and expand its spectrum of activity, resulting in compounds of specific usefulness—for example, in urinary infections (sulfonamides that are highly soluble) or within the gastrointestinal tract (nonabsorbable sulfonamides).
Trimethoprim is a 2,4-diaminopyrimidine and, as such, inhibits the enzyme dihydrofolate reductase, resulting in interference in folic acid and subsequent pyrimidine synthesis in the bacterial cell. Trimethoprim is one of several such compounds synthesized and studied by Hitchings and co-workers in the 1950s and 1960s. The use of trimethoprim as a potentiator of sulfonamide activity was introduced by Bushby and Hitchings2 in 1968. In the subsequent decade, the combination of trimethoprim-sulfamethoxazole (TMP-SMX) was introduced clinically and gained a place in chemotherapy for many infectious diseases. These agents, available in a fixed-drug combination, show true antibacterial synergism against a wide variety of organisms.
Sulfonamides
Structure
The clinically useful sulfonamides are derived from sulfanilamide, which is similar in structure to para-aminobenzoic acid (PABA), a factor required by bacteria for folic acid synthesis (Fig. 33-1). A free amino group at the 4-carbon position is associated with enhanced activity. Increased activity caused by increased PABA inhibition is associated with substitutions at the sulfonyl radical (SO2), which is attached to the 1-carbon, as seen with sulfadiazine, sulfisoxazole, and sulfamethoxazole, all of which are more active than the parent compound, sulfanilamide. The nature of these substitutions determines other pharmacologic properties of the drug, such as absorption, solubility, and gastrointestinal tolerance. Substitutions at the 4-amino group result in decreased absorption from the gastrointestinal tract (e.g., phthalylsulfathiazole).
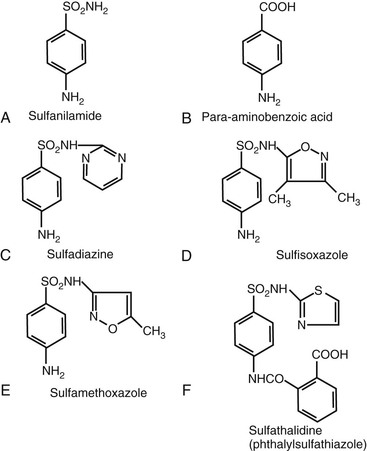
Derivation and Nomenclature
Since the introduction of sulfonamides into clinical medicine, dozens of compounds have been used. Relatively few survive today, however. The various compounds can be classified as (1) short-acting or medium-acting sulfonamides, (2) long-acting sulfonamides, (3) sulfonamides limited to the gastrointestinal tract, or (4) topical sulfonamides. Many branded sulfonamide preparations have been discontinued or replaced by generic products.
Short-Acting or Medium-Acting Sulfonamides
Sulfisoxazole (U.S. Pharmacopeia [USP]; sulphafurazole [British Pharmacopeia]; N′-(3,4-dimethyl-5-isoxazolyl) sulfanilamide is available for urinary infections; sulfamethoxazole USP, N′-(5-methyl-3-isoxazolyl) sulfanilamide is less soluble than sulfisoxazole and yields higher blood levels. It is the sulfonamide most frequently combined with trimethoprim. Sulfadiazine USP (N′-2-pyrimidinylsulfanilamide) is highly active, attains high blood and cerebrospinal fluid levels, and is associated with low protein binding and lower solubility than the previously mentioned drugs.
Short-acting sulfonamides also are available in several combinations. Sulfisoxazole and sulfamethoxazole have been combined with phenazopyridine, a urinary analgesic.
Long-Acting Sulfonamides
Sulfamethoxypyridazine (N′-[6-methoxy-3-pyridazinyl] sulfanilamide) and sulfameter (N′-[5-methoxy-2-pyrimidinyl] sulfanilamide) are no longer available for single-daily-dose therapy because they were associated with hypersensitivity reactions such as Stevens-Johnson syndrome. Neither sulfadimethoxine nor any other long-acting sulfonamides other than sulfadoxine is currently available in the United States.
Sulfadoxine, originally known as sulformethoxine (N′-[5,6-dimethoxy-4-pyrimidyl] sulfanilamide) is a very long-acting sulfonamide that, combined with pyrimethamine, was formerly available in the United States. Sulfadoxine has a half-life of 100 to 230 hours and reaches a peak serum level of 51 to 76 µg/mL 2.5 to 6 hours after an oral dose of 500 mg. Although resistance is noted, this combination is still used in malaria prophylaxis during pregnancy in some countries.3
Sulfonamides Limited to the Gastrointestinal Tract
Sulfaguanidine (N′-[diaminomethylene] sulfanilamide), sulfasuxidine (succinylsulfathiazole [4′-{2-thiazolylsulfamoyl}] succinanilic acid), and sulfathalidine (phthalylsulfathiazole [4′-{2-thiazolylsulfamoyl}] phthalanilic acid) are relatively poorly absorbed from the gastrointestinal tract. They have been used in the past to suppress the susceptible bowel flora before surgery. Salicylazosulfapyridine (sulfasalazine, Azulfidine) is a sulfonamide derivative used to treat ulcerative colitis. This drug is absorbed in its parent form as sulfapyridine, and significant blood levels of this compound are measurable.
Topical Sulfonamides
Mafenide acetate (para-aminomethylbenzene sulfonamide) is available for use in the topical treatment of burns. Its use has been limited, however, by metabolic acidosis caused by carbonic anhydrase inhibition. Silver sulfadiazine has fewer side effects and is used for burns,4 although newer silver compounds are being introduced.5 In these formulations, the sulfonamide acts primarily as a vehicle for release of silver ions that exert an antibacterial effect. Outbreaks of silver-resistant infections in burn units ultimately may limit its usefulness.6 Various combinations of other sulfonamides are available as vaginal creams or suppositories. A variety of ophthalmic ointments and solutions of sulfacetamide sodium USP (a highly soluble sulfonamide) are available for use in the treatment of conjunctivitis caused by susceptible bacteria and as adjunctive therapy for trachoma.
Mechanisms of Action
Although a wide variety of chemical modifications of the sulfonamides have been synthesized, all basically share the same mechanism of action. The sulfonamides are bacteriostatic in that they inhibit bacterial growth by interfering with microbial folic acid synthesis. More specifically, sulfonamides inhibit competitively the incorporation of PABA into tetrahydropteroic acid,7 and they may be incorporated into dihydropteroate.8 Sulfonamides may have a higher affinity for the microbial enzyme tetrahydropteroic acid synthetase than the natural substrate PABA. Richmond9 has suggested that sulfonamides may act on bacterial repressor genes or by feedback inhibition to decrease formation of new enzymes. The ultimate result of decreased folic acid synthesis is a decrease in bacterial nucleotides, with subsequent inhibition of bacterial growth.
Antimicrobial Activity in Vitro
Sulfonamides exhibit in vitro inhibitory activity against a broad spectrum of gram-positive and gram-negative bacteria and Mycobacterium, Actinomyces, Chlamydia, Plasmodium, and Toxoplasma species and some fungi. The in vitro antimicrobial sensitivity of sulfonamides is influenced strongly by the size of the inoculum and the composition of the test medium. High concentrations of PABA and thymidine inhibit sulfonamide activity.
Antimicrobial Resistance
Resistance to sulfonamides is widespread and increasingly common in community-acquired and nosocomial strains of bacteria, including streptococci, staphylococci, Enterobacteriaceae, Neisseria spp., and Pseudomonas spp. Cross-resistance among different sulfonamides is common. In recent decades, sulfonamide resistance in Enterobacteriaceae and gram-positive bacteria has increased substantially.10–12
Organisms may develop a structural change in dihydropteroate synthetase that produces an enzyme with lowered affinity for sulfonamide. PABA overproduction has been implicated in resistant strains of Neisseria gonorrhoeae and Staphylococcus aureus; altered dihydropteroate synthetase has been found in strains of Escherichia coli.13 Resistance also may be mediated by plasmids that code for the production of drug-resistant enzymes, such as dihydropteroate synthetase,14 or decrease bacterial cell permeability to sulfonamides. Plasmid transfer can occur in the gastrointestinal tract and in vitro and has been seen especially with multiple species of Enterobacteriaceae.15
Transformational exchanges of dihydropteroate synthetase among Neisseria spp. raise the specter of wider dissemination of sulfonamide resistance by chromosomal and plasmid genes. More than one resistance mechanism may be operating simultaneously.16
Plasmid-mediated sulfonamide resistance has increased greatly in more recent years, often in conjunction with trimethoprim resistance. Salmonella resistance to sulfonamides has also increased, often in conjunction with resistance to other antibiotic classes. A recent U.S. survey showed 32% of 1118 Shigella isolates were sulfonamide resistant.11 The increase in sulfonamide-resistant Haemophilus ducreyi in Asia and Africa has been associated with a plasmid related to plasmids found in Enterobacteriaceae.17 More than 20% of 442 clinical isolates of Neisseria meningitidis from an international survey were resistant to sulfonamide.18 Bacterial resistance to sulfonamides has increased among human immunodeficiency virus (HIV)-infected patients during the era of increased use of TMP-SMX prophylaxis.
Pharmacology
Routes of Administration
Sulfonamides are usually administered orally. Sulfacetamide is available as ophthalmic preparations; silver sulfadiazine and mafenide acetate are applied topically in burn patients and are associated with significant absorption of sulfonamide percutaneously. Vaginal preparations are available for topical application.
Absorption
Most of the short-acting and medium-acting sulfonamides are absorbed rapidly and almost completely in the nonionized state from the small intestine and stomach. Compounds with N-1 substitutions are absorbed poorly, as are more acidic compounds (e.g., phthalylsulfathiazole; see Fig. 33-1F). Topical sulfonamides are absorbed and may be detectable in blood.
Distribution
The sulfonamides are generally well distributed throughout the body, entering the cerebrospinal fluid and synovial, pleural, and peritoneal fluids with concentrations approaching 80% of serum levels. Blood and tissue levels are related to the degree of protein binding (Table 33-1) and lipid solubility. Sulfonamides administered in pregnancy readily cross the placenta and are present in the fetal blood and amniotic fluid.
TABLE 33-1
Blood Levels, Cerebrospinal Fluid Levels, Plasma Half-life, and Protein Binding of Some Sulfonamides
| DRUG | PEAK BLOOD LEVEL* (mg/mL) | LEVEL IN CSF (%) | PLASMA HALF-LIFE (hr) | PROTEIN BINDING (%) |
| Sulfadiazine | 30-60 | 40-80 | 17 | 45 |
| Sulfisoxazole | 40-50 | 30-50 | 5-6 | 92 |
| Sulfamethoxazole | 80-100 | 25-30 | 11 | 70 |
| Sulfadoxine | 50-75 | 20-30 | 100-230 | 80-98 |

* Approximate free sulfonamide level after a 2-g oral dose.
CSF, cerebrospinal fluid.
Metabolism and Excretion
Acetylation and glucuronidation occur in the liver, and free and metabolized drug appears in the urine. Glomerular filtration is probably a route of excretion, although partial reabsorption and active tubular secretion are also involved, especially at low creatinine clearance rates. Urinary excretion is more rapid for sulfonamides with low pKa values (e.g., sulfamethizole, sulfisoxazole), and alkalinization of the urine increases excretion by this route. Plasma half-lives vary widely; they are related inversely to lipid solubility and directly to pKa values but are not related clearly to the degree of protein binding. Small amounts of sulfonamides are found in bile, human milk, prostatic secretions, saliva, and tears.19
Sulfamethoxazole is primarily metabolized by the CYP2C9 hepatic enzyme system, but also by CYP3A4, and the substrate inhibits the activity of CYP2C9.20 Excretion may be decreased in older patients, especially those with decreased creatinine clearance. Sulfadoxine half-life and area under the concentration versus time curve (AUC) are reduced, and clearance is increased during pregnancy.21
Protein Binding and Blood or Tissue Levels
Sulfonamides are bound variably and not irreversibly to plasma albumin, and the bound drug is inactive (see Table 33-1). Levels obtainable in cerebrospinal fluid and other body fluids are related inversely to the degree of protein binding. The amount of free drug in plasma is related directly to the pKa.
Toxicity and Adverse Reactions
Sulfonamides can cause nausea, vomiting, diarrhea, rash, fever, headache, depression, jaundice, hepatic necrosis, drug-induced lupus,22 and a serum sickness–like syndrome. Sulfadiazine used in excessively high doses is associated with crystalluria and tubular deposits of sulfonamide crystals. These complications can be minimized by maintenance of high urine flow and alkalinization of the urine. Tubular necrosis, interstitial nephritis, and necrotizing angiitis may be associated rarely with sulfonamide sensitivity.
More serious adverse reactions caused by sulfonamides may include acute hemolytic anemia sometimes related to a deficiency in erythrocyte glucose-6-phosphate dehydrogenase (G6PD), aplastic anemia, agranulocytosis, thrombocytopenia, and leukopenia.
Sulfonamides administered during the last month of pregnancy compete for bilirubin-binding sites on plasma albumin and may increase fetal blood levels of unconjugated bilirubin, increasing the risk of kernicterus. Also, because of the immature fetal acetyltransferase system, blood levels of free sulfonamide may be increased, further adversely affecting the risk of kernicterus.
Significant hypersensitivity reactions can occur with sulfonamides administered via any route. The most important of these reactions are erythema nodosum, erythema multiforme (including Stevens-Johnson syndrome), fixed-drug eruption, vasculitis similar to periarteritis nodosa, and anaphylaxis. One report has suggested that cutaneous reactions, including toxic epidermal necrolysis, may be related to an inherited constitutional defect in detoxification of metabolites.23 Locally applied sulfonamides (e.g., to skin) may be associated with any of these adverse reactions.
Many HIV-infected patients who have adverse reactions to sulfa drugs can be desensitized by gradual dose escalation or may tolerate rechallenges without severe adverse reactions.24 Patients who are not desensitized successfully to sulfa drugs have tolerated changing their regimen to dapsone, with or without pyrimethamine.
Drug Interactions
Sulfonamides may displace from albumin-binding sites drugs such as warfarin, increasing the effective activity of the displaced drug. Anticoagulant dosage should be reduced during sulfonamide therapy. Sulfonamides also displace methotrexate from its bound protein, increasing methotrexate toxicity. An increased hypoglycemic effect of chlorpropamide and tolbutamide may occur during sulfonamide therapy, possibly because of the same mechanism or structural similarities. Sulfonamides may compete for binding sites with some anesthetic agents such as thiopental, and reduced barbiturate doses might be necessary. Sulfonamides may potentiate the action of some thiazide diuretics, phenytoin, and uricosuric agents. Conversely, sulfonamides themselves can be displaced from binding sites by indomethacin, phenylbutazone, salicylates, probenecid, and sulfinpyrazone, resulting in increased sulfonamide activity. Cyclosporine levels may be reduced by sulfonamides. Oral contraceptive failure during sulfonamide therapy has been noted rarely.25
The activity of sulfonamides may be decreased by procaine and other local anesthetics derived from PABA. Methenamine compounds should not be used with sulfonamides because of the formation of insoluble urinary precipitates. Sulfonamides may decrease protein-bound iodine and 131I uptake and may produce false-positive Benedict test results for urine glucose and false-positive sulfosalicylic acid test results for urine proteins.
Major Clinical Uses
Sulfonamides are used in the treatment of acute urinary tract infections. Increasing resistance has diminished their effectiveness. Because of widespread resistance of uropathogenic E. coli, the choice of therapy should be based on community prevalence of susceptible infecting organisms (see Chapter 74).
Sulfonamides are also effective for the treatment of infections caused by Nocardia asteroides, although resistance may be increasing.26 Therapy must include 4 to 6 g or more daily after a loading dose of 4 g and should be continued for 4 to 6 months or longer, if necessary (see Chapter 255). Sulfonamides may be useful in combination with other antimycobacterial drugs for the management of infections caused by some atypical mycobacteria (see Chapter 254).
Sulfonamides have been used to treat toxoplasmosis in patients with or without acquired immune deficiency syndrome (AIDS) and chloroquine-sensitive or chloroquine-resistant Plasmodium falciparum malaria (with pyrimethamine). Studies have noted the successful prophylactic role of sulfadoxine-pyrimethamine treatment in pregnant women,3 but increased resistance has been reported. The optimal treatment for toxoplasmic encephalitis is the combination of pyrimethamine plus sulfadiazine or, for patients intolerant to sulfonamides, pyrimethamine plus clindamycin. In most studies, both regimens seem equally efficacious.27 The pyrimethamine-sulfadiazine regimen begins with a 4- to 8-week induction course of pyrimethamine (100 to 200 mg once daily for 1 day, followed by 50 to 75 mg once daily) plus sulfadiazine (1 to 2 g four times daily). Leucovorin (5 to 50 mg once daily) is administered to prevent pyrimethamine-associated folinic acid deficiency. Sulfadiazine may be given twice daily (2 g), even during induction therapy.28 Sulfonamide desensitization has been effective for patients with cerebral toxoplasmosis and allergies to sulfonamides. High-dose trimethoprim-sulfamethoxazole (see later) is an effective alternate for AIDS-related cerebral toxoplasmosis, but pyrimethamine-sulfadiazine is preferable.27 Melioidosis, dermatitis herpetiformis, lymphogranuloma venereum, and chancroid have responded to sulfonamides. Nongonococcal urethritis caused by Chlamydia, but not that caused by Ureaplasma urealyticum, responded to sulfonamide therapy (see Chapters 182 and 186). Sulfasalazine is used in the treatment of inflammatory bowel diseases.29 Currently, sulfonamides are used most frequently in combination with trimethoprim (see later).
Trimethoprim
Structure and Derivation
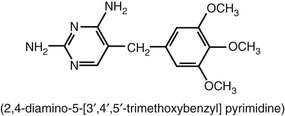
Trimethoprim is a 2,4-diamino-5-(3′,4′,5′-trimethoxybenzyl) pyrimidine (Fig. 33-2). This drug was synthesized by Bushby and Hitchings2 as a dihydrofolate reductase inhibitor thought to potentiate the activity of sulfonamides by sequential inhibition of folic acid synthesis. In the United States, trimethoprim is available as a single agent and in combination with sulfamethoxazole (co-trimoxazole; see later). Trimethoprim has antibacterial activity of its own.
Mechanism of Action
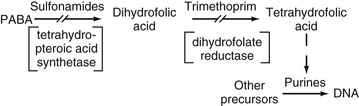
Trimethoprim owes its activity to powerful inhibition of bacterial dihydrofolate reductase, which is the enzyme step after the step in folic acid synthesis blocked by sulfonamides. Trimethoprim is 50,000 to 100,000 times more active against bacterial dihydrofolate reductase than against the human enzyme. Trimethoprim interferes with the conversion of dihydrofolate to tetrahydrofolate, the precursor of folinic acid and ultimately of purine and DNA synthesis (Fig. 33-3). The sequential blockage of the same biosynthetic pathway by sulfonamides and trimethoprim results in a high degree of synergistic activity against a wide spectrum of microorganisms. Humans do not synthesize folic acid but require it in their diet, and human purine synthesis is not affected significantly by the enzyme inhibition of trimethoprim.30
Antimicrobial Activity
Trimethoprim is active in vitro against many gram-positive cocci and most gram-negative rods, except for Pseudomonas aeruginosa and Bacteroides spp. Treponema pallidum, Mycobacterium tuberculosis, Mycoplasma spp., and most anaerobes are resistant. Thymidine inhibits the in vitro activity of trimethoprim, but the addition of thymidine phosphorylase or 5% lysed horse blood to Mueller-Hinton medium or other sensitivity media removes this inhibition. The minimal inhibitory concentration (MIC) varies considerably with the medium used. Trimethoprim alone has good in vitro activity against H. influenzae; a recent survey has shown that 76% of 978 strains remain susceptible, but higher resistance rates are reported elsewhere.31 Streptococcus pyogenes MICs have been reported at less than or equal to 2 µg/mL.32
Resistance to Trimethoprim
Bacteria may develop trimethoprim resistance by several mechanisms, which can be chromosome or plasmid mediated. Clinical resistance has increased. A decrease in the ratio of strains resistant to sulfamethoxazole and trimethoprim compared with strains resistant only to trimethoprim may reflect an increase in independent trimethoprim resistance, which might be a useful monitoring parameter in hospitals.33
Trimethoprim resistance may be caused by changes in cell permeability, loss of bacterial drug-binding capacity, and overproduction of or alterations in dihydrofolate reductase. Clinically, the most important mechanism is plasmid-mediated dihydrofolate reductases that are resistant to trimethoprim.34 Distinctive dihydrofolate reductases have been described in Enterobacteriaceae, P. aeruginosa, H. influenzae, Streptococcus pneumoniae, S. aureus, and Campylobacter spp. (chromosomally resistant to high trimethoprim concentrations). They are frequently plasmid mediated and may be disseminated by highly mobile transposons (e.g., Tn7) with wide host species ranges. Outbreaks caused by trimethoprim-resistant conjugative plasmids have been noted in western and eastern Europe, eastern Asia, South America, and the United States.35 Many of the outbreaks occurred in immunocompromised hosts, associated with resistance to multiple other antibiotic groups.
Variable local increases in trimethoprim resistance, particularly among Enterobacteriaceae, have been reported. A recent survey found trimethoprim resistance in 15% to 19% of E. coli uropathogens.36 Whether the clinical use of trimethoprim alone in some countries has resulted in increasing resistance to TMP-SMX is unclear.
Pharmacology
Routes of Administration
Trimethoprim is available as 100-mg tablets for oral use. Trimethoprim is absorbed readily and almost completely from the gastrointestinal tract. Peak serum levels appear 1 to 4 hours after ingestion of 100 mg and approach 1 µg/mL. The co-administration of sulfamethoxazole does not affect the rate of absorption or serum levels of trimethoprim.
Distribution
Trimethoprim is distributed widely in tissues and may appear in kidney, lung, and sputum in higher concentrations than in plasma and in bile, saliva, human breast milk, and seminal fluid.37 Trimethoprim is also found in prostatic fluid at two to three times the serum concentration, but lower levels may be present in patients with chronic prostatitis. Cerebrospinal fluid concentrations are about 40% of serum levels.
Metabolism and Excretion
Approximately 60% to 80% of an administered dose of trimethoprim is excreted in the urine via tubular secretion within 24 hours. The remainder of the drug is excreted by the kidney in one of four oxide or hydroxyl derivatives. The urinary metabolites are bacteriologically inactive.38 Trimethoprim is also a major substrate of CYP2C9 and a minor substrate of CYP3A4. Its metabolism inhibits CYP2C8 and CYP2C9.20 Trimethoprim is also excreted in the bile. The serum half-life ranges from 9 to 11 hours in healthy subjects and is prolonged in patients with renal insufficiency. In contrast to sulfamethoxazole, the excretion rate of trimethoprim is increased with acidification of the urine, and serum protein binding (65% to 70%) does not decrease significantly with increasing degrees of uremia. Urine concentrations in healthy subjects (60 to 1000 µg/mL) are usually greater than the MIC of many urinary pathogens.
Clinical Use
The primary use of trimethoprim alone is in the prevention of recurrent uncomplicated urinary tract infections in women with structurally normal anatomy.39 Trimethoprim has also been successful in the treatment of acute urinary tract infections at a dose of 100 mg twice daily,40 but increasing resistance threatens its usefulness.34
Trimethoprim-Sulfamethoxazole
Mechanism of Action and Antimicrobial Activity
Potentiation of the action of trimethoprim is seen in combination with sulfamethoxazole. The combination of trimethoprim and sulfamethoxazole (TMP-SMX, co-trimoxazole) is active in vitro against many isolates of S. aureus, including the prevalent community-associated methicillin-resistant S. aureus (MRSA) clone.41 In vitro TMP-SMX activity is reported for S. pyogenes, S. pneumoniae (although resistance is increasing in patients receiving TMP-SMX prophylaxis42), Moraxella catarrhalis, E. coli, Proteus mirabilis, Burkholderia cepacia, Burkholderia pseudomallei,4,43 Yersinia enterocolitica (but resistance reported), N. gonorrhoeae (resistance rates higher than 20% reported44), and Stenotrophomonas maltophilia. TMP-SMX–resistant strains of S. maltophilia are increasing and may occur in severely ill patients with serious morbidity.45 Of pneumococci carried in the nasopharynx of children treated for malaria with a sulfonamide or TMP-SMX, 50% were resistant to TMP-SMX.46 A study from Finland has documented an increasing prevalence of TMP-SMX resistance in S. pneumoniae, M. catarrhalis, and H. influenzae and found that regional use of TMP-SMX was associated with increasing resistance among pneumococci.47 Variable TMP-SMX resistance has been reported in penicillin-susceptible and nonsusceptible pneumococci.48 A large Finnish study of more than 20,000 S. pneumoniae isolates correlated TMP-SMX resistance with increasing use.49 A more recent study among HIV-infected South African patients with invasive pneumococcal infections reported that TMP-SMX prophylaxis was associated with pneumococcal nonsusceptibility to TMP-SMX, as well as to rifampin and penicillin.42 The Etest might not be reliable for the determination of TMP-SMX–resistant pneumococci. A wide range of TMP-SMX susceptibilities among Salmonella and Shigella spp. has been reported.
Variable bactericidal effects have been noted when enterococci are tested against TMP-SMX, but TMP-SMX is not clinically useful. However, one report has suggested that standard in vitro testing might not predict clinical outcome and might underestimate the effectiveness of trimethoprim alone in enterococcal urinary tract infections.50 The susceptibility of Enterobacteriaceae may vary greatly among locations and within the same location from year to year because of the spread of trimethoprim-resistant plasmids and transposons. Almost all strains of P. aeruginosa are resistant in vitro to TMP-SMX.
Trimethoprim combined with sulfamethoxazole or dapsone has been effective in the treatment of Pneumocystis jirovecii pneumonia in immunocompromised patients (see later). Listeria monocytogenes, M. catarrhalis, and atypical mycobacteria have been shown to be susceptible to TMP-SMX, but variable rates of resistance are reported.
The optimal ratio for in vitro synergism of TMP-SMX in combination is 1 : 20, but this ratio does not always obtain in vivo. The synergism seen depends on the sensitivity of the organism to each drug.
Trimethoprim-Sulfamethoxazole Resistance
Concomitant trimethoprim and sulfonamide resistance may limit the usefulness of TMP-SMX against some strains of methicillin-resistant S. aureus and Staphylococcus epidermidis. However, the most commonly circulating CA-MRSA clone (USA300) is usually susceptible to TMP-SMX.41,51 In HIV-infected patients, TMP-SMX use has been associated with TMP-SMX resistance among S. aureus and Enterobacteriaceae.52 There is limited evidence that TMP-SMX selects for resistance to other antibiotic classes.53 Multiple drug resistance is common now in E. coli isolated from the urinary tract; TMP-SMX resistance rates approach 20% to 25% in the United States.54,55
Permeability changes may occur in the bacterial cell and result in resistance to trimethoprim and sulfonamides. Thymine-requiring auxotrophs may also account for clinically significant resistance to both drugs. These mutants lack thymidylate synthetase and are probably less virulent than sensitive strains.34 Many Shigella spp. are resistant, and resistance is increasing among Salmonella spp.,56 although most Salmonella typhi strains retain susceptibility. P. jirovecii may also develop sulfonamide and trimethoprim resistance mutations in the course of therapy of immunocompromised HIV-infected patients, resulting in treatment failure.57
Trimethoprim is also available in fixed combination with sulfamethoxazole in a ratio of 1 : 5 for oral use (trimethoprim, 80 mg; sulfamethoxazole, 400 mg). Double-strength and quarter-strength pediatric tablets are available, as is an oral suspension containing 40 mg of trimethoprim and 200 mg of sulfamethoxazole per 5 mL. IV trimethoprim (16 mg/mL) plus sulfamethoxazole (80 mg/mL) is available. When administered intravenously, 10 mL or 160 mg of trimethoprim (with 800 mg of sulfamethoxazole) produces a peak serum trimethoprim concentration of 3.4 µg/mL in 1 hour. After repeated doses, the peak trimethoprim concentration may approach 9 µg/mL.38 Similar peak levels may be reached with oral therapy, but at 2 to 4 hours after administration.58
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


