two GATA-binding consensus sequences.30 The endothelial cell-specific expression appears to be regulated by a repressor-derepressor mechanism and includes both an NF1-binding site and an Oct-1-binding site.30,31,32,33,34 A more complex mode of transcriptional regulation through vascular bed-specific signaling pathways has been recently defined. A 2,200 base pair 5′ flanking sequence together with the first intron and exon direct expression to microvascular endothelial cells of skeletal muscle and heart.35 Recently, cell type-specific regulation of vWF expression by the E4BP4 transcriptional repressor has been identified.36 Within the heart, a cardiomyocyte-dependent signaling pathway through platelet-derived growth factor (PDGF) has been defined.37 Several single-nucleotide polymorphisms at nucleotides -1793, -1234, -1185, and -1051 have been associated with plasma vWF:Ag levels.38,39 Thus, vWF gene is regulated by both cell-specific elements as well as the environment in which these cells are growing.31,40
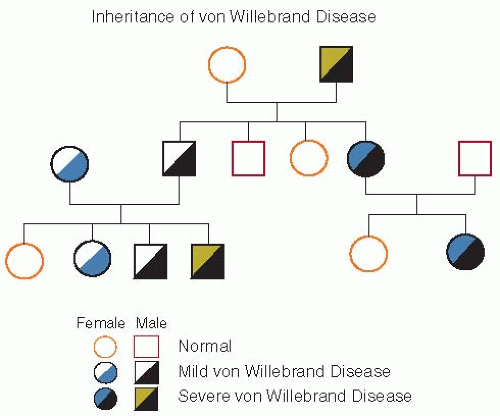 FIGURE 13.1 The autosomal inheritance of von Willebrand disease (vWD) is illustrated in this figure. Because low von Willebrand factor (vWF) has a frequency of 1% to 2%, some of these individuals are asymptomatic. Those with symptoms are said to have mild type 1 vWD. Patients with type 3 vWD are homozygous and inherit null alleles from each parent. (Used by permission, Montgomery RR.) |
Table 13.1 Terminology | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
and is identical to the protein sequence derived from the cDNA sequence with a molecular weight of 225,663 for just the amino acid sequence of the mature vWF.3,4,5,49 The carbohydrate component is estimated to add approximately 10% to 15% to the molecular mass; thus, the true molecular weight of the monomer is approximately 255,000, and its migration in polyacrylamide gel underestimates its true molecular weight.
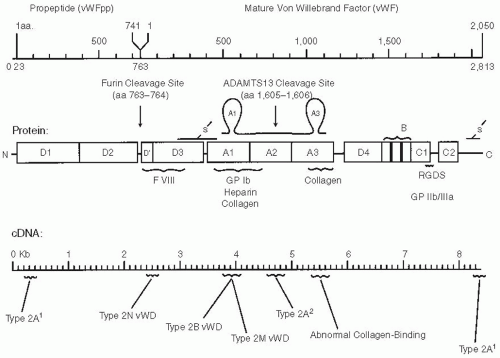 FIGURE 13.2 The von Willebrand factor (vWF) protein is initially synthesized as a 2,813-amino acid pro-vWF molecule whose synthesis is directed by an 8.5-kb RNA. The pre-pro-vWF is composed of a 22-amino acid signal peptide, a 741-amino acid propeptide (vWFpp), and a 2,050-amino acid mature vWF monomer. The pro-vWF is composed of A, B, C, and D repeats. Various functional domains have been identified and contain the sites of interaction with factor VIII, platelet glycoprotein GPIb, GPIIb/IIIa, collagen, and heparin. On the basis of these functional domains, various genetic mutations have been mapped to discrete regions of the cDNA as illustrated. Two protease domains are illustrated: furin cleavage site (cleaves the vWFpp from vWF) and ADAMTS13 cleavage site that cleaves the A2 domain. This ADAMTS13 cleavage is increased by some vWF mutations that cause type 2A vWD caused by increased vWF proteolysis. Cleavage by ADAMTS13 is absent in patients with thrombotic thrombocytopenic purpura (TTP) either because of a deficiency of the enzyme (hereditary TTP) or an autoantibody that blocks its cleavage of vWF (acquired or sporadic TTP). Type 2 mutations are generally located in specific regions along the vWF protein. Types 2A, 2B, and 2M are primarily located within exon 28 that encodes for the A1 and A2 regions of vWF. The two different types of 2A are those that have increased proteolysis 2A2 and those with abnormal multimer synthesis 2A1. Mutations with abnormal collagen binding are clustered in the region encoding the A3 domain. (Used by permission, Montgomery RR.) |
vWD, in which there is a reduction in multimeric size, often have more severe bleeding symptoms than individuals with a similar reduction in protein concentration but normal multimeric size (type 1 vWD); thus, in vivo, multimeric size may correlate with function.55
cysteine motifs similar to disulfide isomerase.95 The propeptide was shown to act as an oxidoreductase, thus promoting the multimerization of vWF in the Golgi apparatus.96 Additional studies identified two amino acids in the D3 domain, Cys-1099 and Cys-1142, that participate in this process.76
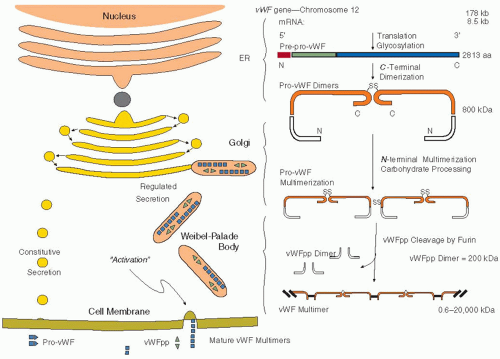 FIGURE 13.3 The sites of cellular biosynthesis are represented schematically in this illustration of endothelial cell biosynthesis. The von Willebrand factor (vWF) protomer is synthesized as a pre-pro-vWF dimer that is a C-terminal disulfide-bonded dimer within the endoplasmic reticulum (ER). The 22-amino acid signal peptide is then cleaved. The carbohydrate is then processed into complex sugars within the Golgi compartment. N-Linked multimerization by virtue of the selfassociation of the vWF propeptide (vWFpp) occurs in the acidic compartment of the late-Golgi, and the vWFpp is cleaved from the mature vWF by furin. A pH-dependent association between vWFpp and vWF enables the packaging of both proteins into storage granules termed Weibel-Palade bodies. After activation of the endothelial cell, the regulated secretion of these proteins occurs, but at the neutral pH in plasma, there is no longer an association of vWFpp with vWF. A similar pathway is assumed to occur in megakaryocytes, but the α-granule contains more different types of proteins. In the absence of vWF, the Weibel-Palade body is not formed, but the α-granule is still present. (Used by permission, Montgomery RR.) |
granular storage of vWF is reestablished.80,103 The propeptide contains the necessary sequence or conformation for sorting to storage granules and secondarily cotraffics mature vWF multimers through noncovalent association.80 The nature of the noncovalent association between propeptide and mature vWF has been further defined. Two amino acids, arginine 416 within the propeptide and threonine 869 in the mature vWF protein are essential for noncovalent association and regulated storage of vWF.104 The putative sorting signal for the propeptide may been located in the D2 domain. By using a series of propeptide truncation cDNA constructs (FIGURE 13.4), the sorting signal has be localized within amino acids 387 to 545 of the propeptide. The sorting signal within the vWF propeptide has been used to traffick an unrelated, nonsecretory granule protein (C3α) to the regulated storage pathway in AtT-20 cells and endothelial cells.90
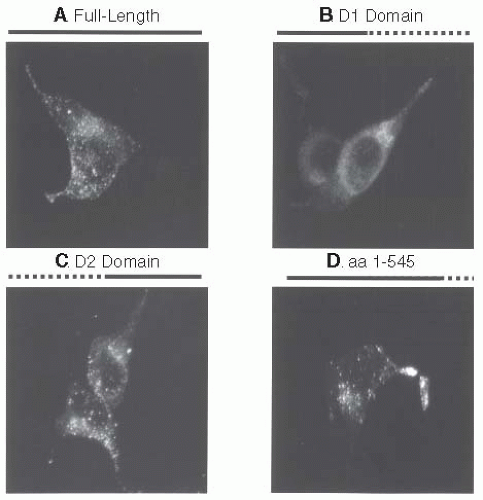 FIGURE 13.4 At T-20 cells were transiently transfected with von Willebrand factor (vWF) propeptide truncation expression plasmids, immunofluorescently stained, and examined by confocal microscopy. A: Cells expressing full-length propeptide demonstrate a granular staining pattern. B: Cells expressing only the D1 domain (amino acids 1 to 386) of the propeptide demonstrate a diffuse staining pattern with no granules detected. C: Cells expressing only the D2 domain (amino acids 386 to 763) of the propeptide demonstrate a granular staining pattern similar to full-length propeptide. D: Granular staining of a propeptide consisting of amino acids 1 to 545. The putative vWF propeptide sorting signal lies in the D2 domain within the region of amino acids 387 to 545. (Used by permission, Montgomery RR and Haberichter SL.) |
unpublished data), P-selectin-containing alpha-granules are still present in platelets and megakaryocytes. Therefore, vWF is required for the biogenesis of the Weibel-Palade body but not the alpha-granule.
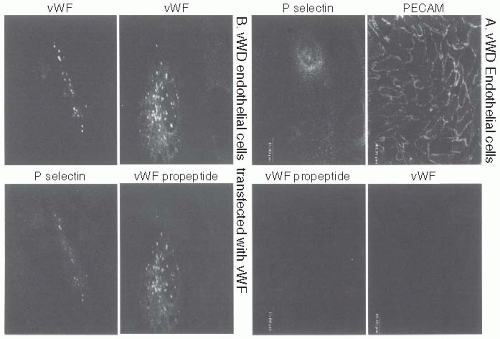 FIGURE 13.5 A: Endothelial cells were harvested from the aortas of dogs with type 3 von Willebrand disease (vWD), cultured, immunofluorescently stained, and examined by confocal microscopy. The cells were platelet/endothelial cell adhesion molecule (PECAM) (CD31)-positive, indicating that a homogenous population of endothelial cells had been cultured as shown. The cells were also found to be vWF-negative as shown. Staining for P-selectin was faint and diffuse with many small granules, most likely lysosomes. The vWF propeptide could not be detected in these cells. Therefore, no apparent Weibel-Palade bodies were detected in the canine vWD aortic endothelial cells. B: The canine vWD aortic endothelial cells were transiently transfected with a vWF expression plasmid. Immunostaining of cells expressing vWF demonstrated a granular distribution of vWF and vWF propeptide as shown. Dual staining for vWF and P-selectin revealed that the vWF was colocalized with P-selectin in granules. Therefore, in the absence of vWF, no Weibel-Palade bodies are formed in endothelial cells. However, vWF expression induces the formation of Weibel-Palade bodies, and the Weibel-Palade body distribution of endogenous P-selectin is reestablished. (Used by permission, Montgomery RR and Haberichter SL.) |
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


