Chapter 4 Strategic Use of Antiretroviral Therapy
Over the last decade, highly active antiretroviral therapy (HAART) has revolutionized the care of human immunodeficiency virus (HIV) infected patients. Advances in the understanding of HIV pathogenesis, the routine use of viral load and resistance testing in clinical practice, and the availability of over 23 Food and Drug Administration (FDA) approved antiretroviral (ARV) agents (Table 4-1) have created the promise for most HIV-infected patients to live, and live well, for decades such that HIV ultimately has little impact on the duration or quality of their life.1–5 Persistent, high-level viral replication is the driving force of HIV pathogenesis, with up to 10 billion virions produced in an infected individual each day.6–9 Viral load measurements enable the clinician to determine the degree to which an ARV therapeutic regimen is working and, more importantly, when the regimen is failing.10–14 This allows therapy to be switched at the time of ARV failure rather than at the time of clinical failure. Once a regimen begins to fail, clinicians can utilize resistance test information to choose from dozens of potential alternative regimens, utilizing both existing approved drugs and experimental therapeutic agents with novel mechanisms of action now in development.15 Through the contributions of all of these developments, clinicians have the potential to achieve long-term clinical benefits by keeping the viral load as low as possible for as long as possible.
Table 4-1 ARV Drugs Approved for Use in the United States as of 2006
| Year Approved | Agent (Trade Name) |
|---|---|
| 1987 | Zidovudine (retrovir) |
| 1991 | Didanosine (videx) |
| 1992 | Zalcitabine (hivid) |
| 1994 | Stavudine (zerit) |
| 1995 | Saquinavir (invirase) |
| Lamivudine (epivir) | |
| 1996 | Indinavir (crixivan) |
| Ritonavir (norvir) | |
| Nevirapine (viramune) | |
| 1997 | Delavirdine (rescriptor) |
| Nelfinavir (viracept) | |
| 1998 | Efavirenz (sustiva) |
| Abacavir (ziagen) | |
| 1999 | Amprenavir (agenerase) |
| 2000 | Lopinavir/ritonavir (kaletra) |
| 2001 | Tenofovir DF (viread) |
| 2003 | Atazanavir (reyataz) |
| Emtricitabine (emtriva) | |
| Enfuvirtide (fusion) | |
| 2004 | Fos-amprenavir (lexiva) |
| 2005 | Tipranavir (aptivus) |
| 2006 | Darunavir (prezista) |
| Miraviroc | |
| 2007 (in expanded access) | Raltegravir |
| Etravirine |
The use of modern ARV therapies has led to a striking reduction in HIV-associated mortality.16–18 Despite these advances, newly recognized toxicities of ARV treatment have begun to limit the long-term benefits of chronic therapy.19–27 Moreover, the durability of the ARV effect is quite variable. Many factors influence the ability to sustain suppression of viral replication, including pharmacokinetic properties of the regimen, tissue penetration, cellular penetration, appropriate intracellular processing, ARV drug history, tolerability of the regimen, adherence, potency of the regimen, and the development of or preexisting presence of resistant virus.15,28–34 To achieve the most durable effect of ARV therapy (ART), clinicians must develop a strategic approach that maximizes the likelihood of success of each given regimen through a thorough understanding of the biology of HIV disease and the principles of ART.
THE BIOLOGY OF HIV INFECTION
Since HIV was identified in 1983, several landmark discoveries have helped elucidate the mechanisms by which HIV causes the immune system dysfunction associated with AIDS. These discoveries are usually linked to the application of newly developed technology in the laboratory. Soon after discovery of the virus, investigators demonstrated the presence of HIV in virtually all tissues of the body, including the brain.35,36 Utilizing p24 antigen assays, an association was made between the level of p24 antigen in plasma and the stage of disease, with higher levels of viremia observed during the time of acute seroconversion and again in the later stages of advanced HIV disease.37–39 Most of the individuals who were asymptomatic with high CD4+ T-lymphocyte counts had no appreciable p24 antigenemia detected. During the late 1980s, utilizing tissue culture techniques investigators were able to titrate the amount of infectious virus in plasma.37,40,41 Much like p24 antigenemia, higher levels of infectious virus in plasma were noted at the time of acute seroconversion and again in later stages of disease.38,39,42 Although the plasma culture technique was more sensitive than the p24 antigen technique, substantial numbers of patients with asymptomatic disease, as well as those on ART, had undetectable levels of infectious virus.37,40,41
Utilizing this information, a picture of HIV pathogenesis emerged whereby the virus established widespread infection early in the course of disease (at the time of seroconversion), stimulating a potent immune system response. During the period of clinical latency, which typically lasts up to 10–12 years, initial models of pathogenesis suggested that viral replication was under effective control by the immune system, only to reappear as high-level viremia during later stages of disease (Fig. 4-1).38,39,42 This model, however, could not explain the slow, yet progressive decline in CD4+ T-lymphocyte counts and immune system function that occurs during the period of clinical latency. With the advent of quantitative polymerase chain reaction (PCR) technology during the early 1990s, the association of HIV replication and immune system destruction was more completely described. Piatak and colleagues were the first to describe the presence of detectable virus at all stages of HIV infection, including the period of clinical latency.12,13 As was demonstrated with p24 antigen and quantitative plasma culture techniques, the highest levels of viral RNA were detected at the time of acute seroconversion and during later stages of disease.12,43 However, lower levels of virus were detected even at the early, asymptomatic stages of the disease, implying that viral replication is a continuous, ongoing process even during the period of clinical latency.
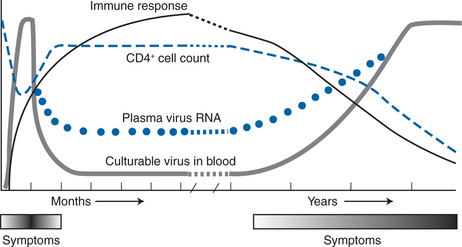
Figure 4-1 Natural history of HIV-1 infection over time.
Modified from Saag MA, Holodniy M, Kuritzkes DR, et al. HIV viral load markers in clinical practice. Nature Med 2:625, 1996. Copyright 1996 Macmillan Magazines Limited.
The application of viral load testing to determine the activity of ARV therapeutic regimens led to the opportunity to define further the nature of HIV replication in vivo. Even with the use of relatively weak ARV regimens, such as zidovudine monotherapy, an 80% (0.9 log) reduction in viral load was noted within 1 week of the initiation of therapy, with a relatively symmetrical return to baseline within 1 week after discontinuing treatment.44 Based on these observations, it was apparent that viral replication was not only continuous but also quite rapid.45 However, it was not until the dramatic responses to HIV protease and non-nucleoside reverse transcriptase (RT) inhibitors were observed that the magnitude of the rate of viral replication in vivo was fully appreciated.
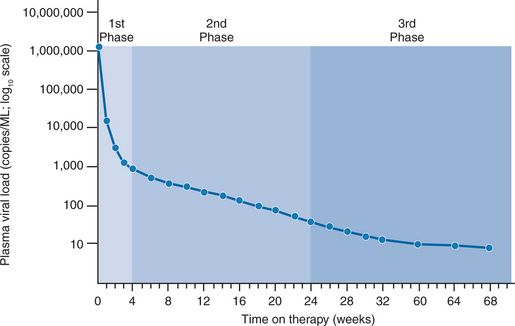
In separate reports, Wei et al and Ho et al quantitated the rapid turnover of HIV in vivo, demonstrating the production and clearance of up to 10 billion virions each day (∼400 million virions per hour).6,7 The half-life of virions in the circulation is estimated to be 1–2 h or less. When new rounds of viral replication are blocked by ART, the amount of measurable virus drops by more than 99% within 2–7 days of initiating therapy. The estimated life cycle (or generation time) of HIV, which represents the time from release of a virion until it infects another cell resulting in the release of new progeny, is estimated to be 1–2 days (Fig. 4-2).8,46,47 These data were generated by observing the rapid decay in viral load over the first few weeks of potent therapy and represent the contribution of the acutely infected cells in the host, which have a half-life of 1–2 days. These cells represent more than 99% of the daily virus production. The remaining production of virus comes from a longer-lived population of cells that contribute to the slower, ‘second phase’ decay of plasma viremia (Fig. 4-3).47,48 Therefore within 8–12 weeks after initiation of potent therapy, plasma HIV RNA levels generally fall below 400 copies/mL of plasma. It usually takes several weeks longer to reach undetectable levels when utilizing ultrasensitive virologic techniques (limit of detection ∼5–50 copies/mL). Most viral replication takes place in lymphoid organs, where most of the CD4+ T lymphocytes reside.28,49–54 The detection of virus in plasma, as measured for example by the plasma ‘viral load’, represents spillover of virus from the site of production (lymphatic tissue) into the bloodstream where it can be readily detected. In addition to the gradual reduction in absolute CD4+ T lymphocytes over time, loss of lymphoid architecture within lymph nodes also occurs and is believed to contribute to the relative lack of immune system efficiency at later stages of the disease.49,50 Although most newly formed virions do not result in infection of a neighboring cell, on average several million new CD4+ T lymphocytes are infected each day.7,46 This high-level, continuous production of virus and subsequent infection of new CD4+ T lymphocytes helps explain how the CD4+ cells decline and immune system destruction occur during the time of clinical latency.
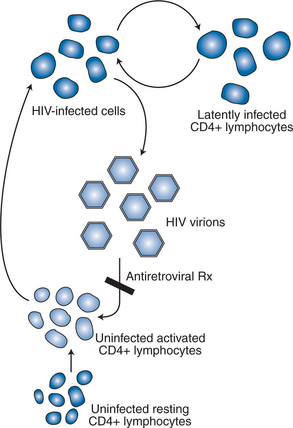
Activated CD4+ T lymphocytes are the principal targets of HIV replication. The estimated life span of an infected CD4+ T lymphocyte is 1–2 days.55 Each infected cell has an instantaneous burst size of ∼4000 copies per cell.28 The calculated burst size per cell is remarkably constant and is independent of stage of disease, plasma viral load, and ARV treatment status.28 Thus the plasma viral load is a direct reflection of the number of infected cells in the body producing virus at any moment in time.
Whereas the instantaneous burst size, defined as the number of measurable virions produced by an infected cell at any given moment, is ∼4000 copies per cell, the effective burst size, defined as the number of viruses produced that result in productive infection of a neighboring cell, at steady state is ∼1 virion per cell (Fig. 4-4). At steady state, each patient establishes an equilibrium between the production and destruction of virus producing cells such that as each productive cell is destroyed or stops producing virus it is replaced by another newly infected, productive cell. Clinically, this equilibrium establishes a relatively stable viral load value over time, defined as the viral ‘set point’.8,56 Mellors and colleagues have demonstrated a direct relation between the viral load, or set point, and the rate of CD4+ T-lymphocyte count decline.57 When viewed in the context of the direct relation between plasma viral load and the number of productively infected cells at any moment in time, these findings are not surprising. Yet, when this concept is applied to individual patients the ability of the plasma viral load to predict the rate of CD4+ T-lymphocyte decline for a specific patient is quite poor, indicating that other factors, as yet unmeasured, are playing a role in causing the loss of T cells in HIV-infected patients.58
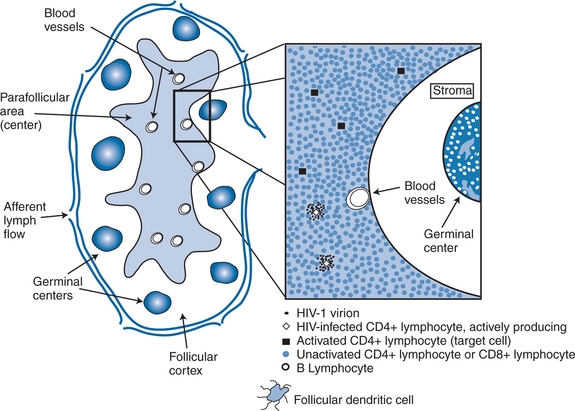
Much attention has focused on how actively producing cells are eliminated.59,60 The leading possibilities are the direct cytopathic effect of the virus (or deleterious effects of virions budding from the cell membrane), virus-induced cell apoptosis, or direct destruction via an effective immune system response. Initial experience with tissue culture growth of the virus supported the concept of a direct cytopathic effect.61 Under the stimulation of phytohemaglutinin and interleukin-2 (IL-2), most virus-infected cells died within several days of becoming infected in tissue culture, often through the development of large syncytia. Syncytia have never been demonstrated in vivo, and so the concept of death of cells via this mechanism remains uncertain. Apoptosis, or programmed cell death, is a natural mechanism for eliminating lymphocytes of the immune system. Viral proteins, or cytokines released in response to viral infection, may trigger the apoptotic pathway in actively producing cells, although the degree to which this occurs is not known. What is clear, however, is the tremendous loss of CD1 T lymphocytes in the gut during the first several days to weeks after initial infection.62–64 Studies in both animals (infected with SIV) and humans with acute HIV infection demonstrate profound loss of immune cells and disruption of normal architecture of the gut in the first several days after infection. These data suggest strong consideration of aggressive use of ART during the period of acute infection.
A large body of evidence supports the role of HIV-specific immune system responses to eliminate virus-producing cells.65–69 The early and rapid development of the HIV quasi-species is a direct consequence of HIV-specific immunity, both cellular and humoral. Neutralizing antibody studies have demonstrated a very dynamic virologic escape phenomena within the first several weeks of HIV infection, with persistent generation of novel viral variants in response to antibody responses over the life of infection.70,71 Enhanced HIV-specific immunologic responses were postulated to be responsible for the lower virologic set points observed among seroconverters who were treated within days to a couple of weeks after the onset of symptoms of their initial HIV infection and had therapy periodically withdrawn.72 High levels of anti-HIV-specific CD4+ T-lymphocyte activity were demonstrated in association with control of replication. This level of CD4+ T-lymphocyte help is typical in ‘long-term nonprogressor’ patients, who naturally control viral infection and do not suffer CD4+ T lymphopenia despite chronic HIV infection. In contrast, when therapeutic withdrawal of treatment was performed in patients with chronic, well-established HIV infection who were not treated within 12–20 weeks of acute infection, viral load values typically rebound back to high levels.73,74 These patients have extremely low or absent HIV-specific CD4+ T-helper lymphocyte responses. Longer-term follow-up of the treated acute seroconversion patients revealed a return to higher viral load set points indicating some loss of the initial immunologic priming from therapy. Of more concern, one patient in the initial intermittent therapy study became superinfected with virus from another patient following sexual exposure while off therapy. This story has created a challenge and some controversy for those working on development of a preventative HIV vaccine.
Taken together, HIV pathogenesis is best depicted as a vicious cycle of production of large numbers of HIV virions that infect activated CD4+ target cells, which in turn produce more viruses that infect additional cells (Fig. 4-5).9 The function of the targeted CD4+ T lymphocytes, ironically, is to create an HIV-specific, coordinated response against the virus that is attacking them. Current thinking supports the concept that cytotoxic CD81 T lymphocytes, under the support of an HIV-specific CD4+ T-lymphocyte response, are responsible, at least in part, for eliminating the active virus-producing HIV-infected cells.68–72,75–78 The HIV-specific CD4+ T-helper lymphocyte responses appear to be lost early (within the first few weeks after infection), perhaps never to return. The continuous infection of other activated CD4+ cells is believed to lead to impairment of the immune responses to other antigens/pathogens, thereby creating potential deficits in the immune system’s response to opportunistic processes. The goal of effective ART therefore is to block, as completely as possible, the ability of the virus to infect uninfected CD4+ T lymphocytes, thereby inhibiting de novo production of virus and, at the same time, preserving immune system competence.61
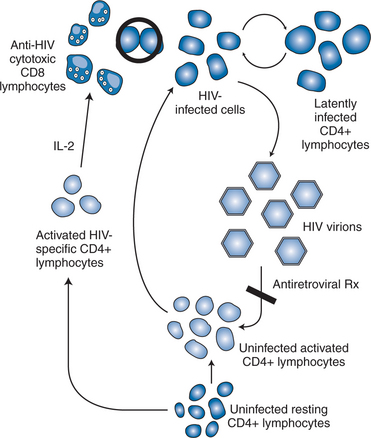
STRATEGIC USE OF ART
Simply stated, the goal of ART is to inhibit completely viral replication in vivo and sustain the effect for as long as possible. Theoretically, if complete suppression is sustained for a long enough time to allow the population of chronically infected cells to decay to extinction, eradication of HIV (or a true cure) is possible. Whether a cure is truly achievable is critical to establishing the foundation of an ARV therapeutic strategy. If cure is indeed possible, all patients should be treated with the most potent agents early and aggressively during the course of their infection, much like the treatment of acute lymphocytic leukemia. If a cure with ART alone is not a realistic possibility, other strategic approaches must be considered, more like the treatment of chronic lymphocytic leukemia.
Whether eradication is possible depends in large part on the life span of longer-lived, chronically infected cells and whether complete (or relatively complete) suppression of viral replication is achievable. About 99% of the plasma viremia is generated by actively producing CD4+ T lymphocytes with a relatively short half-life (1–2 days), the remaining 1% or less of plasma viremia is produced by longer-lived cells that previously were predicted to have an estimated average half-life of 14–28 days, although some of the cells could have a life span as short as 2–4 days or as long as more than 400 days (Fig. 4-2).46,48,79 If the life span of these long-lived cells is 28 days or less and all de novo infection is completely blocked, eradication may be achieved as early as 3–5 years after initiation of therapy46,47; however, if the life span of the longer-lived chronically infected cells is substantially longer (on the order of 400 days) or if latently infected cells (that contain proviral DNA) are able to circulate, multiply or expand, and express virus at a later time, the time required for complete eradication of HIV could be on the order of decades and may not be achievable, even with complete block of de novo rounds of replication sustained over a prolonged period of time.50,67 Data generated during the latter portion of the 1990s demonstrated that the half-life of the chronically infected population of cells was not 14–28 days as originally assumed but, rather, 6 months at the shortest or over 44 months at the longest.29,60,80–82 Thus the time to eradication under the assumption of complete arrest of all de novo infection now ranges from 12 to well over 60 years of continuous, maximally effective ART.
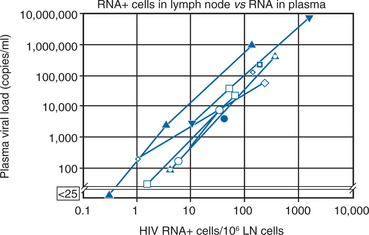
As indicated above, the concept of eradicating HIV from an infected individual requires chronically infected cells to have a relatively short half-life but also requires complete block of all new rounds of de novo infection and this block must be sustained for a long enough time to allow all chronically infected, productive cells within the body to be destroyed or die off. To achieve eradication, inhibitory levels of ART must be maintained above inhibitory concentrations within all body compartments and within all susceptible target cells, ideally on a continuous basis. Thus a ‘cure’ for HIV infection assumes complete penetration of ARV drugs into the cells in all compartments of the body where the virus is replicating and strict adherence to the regimen for a period long enough to allow the infected cells to die off. An important issue in the discussion of a potential ‘cure’ relates to the relation of ‘undetectable’ levels of plasma virus to complete suppression of viral replication. ‘Undetectable’ levels of HIV in plasma are a function of a laboratory assay; complete suppression is a biologic phenomenon. All too often patients and clinicians assume that achieving an undetectable level of virus is synonymous with complete suppression. Even when plasma viral load values are less than a theoretical 1 log10 copy/mL, an estimated 75 000–150 000 cells are still producing virus in lymphoid tissue (Fig. 4-6).28 Zhang et al reported evidence of genetic drift in the envelope region in virus isolated from a patient who was treated at the time of seroconversion and sustained levels of less than 50 copies/mL for more than 20 months, supporting the notion of ongoing low-level replication when plasma viral load values are undetectable.29 Therefore low-level viral replication appears to be ongoing even in the face of potent, yet incomplete viral suppression.59 This implies that breakthrough viremia may ultimately occur in any given patient with sustained plasma HIV RNA levels below 50 copies/mL, although the likelihood of such breakthrough is a function of the probability of emergent resistant virus in the face of strong selective drug pressure (as discussed in the following section). Inevitably, more sensitive virologic assays will become commercially available and help narrow the gap between ‘undetectable’ and ‘complete suppression’. At the present time, however, it does not appear that currently available ARV agents, even when used in combination, are potent enough to achieve and maintain truly complete suppression. Thus a gap continues to exist between the ideal goal of ART (defined as complete suppression of de novo infection) and the achievable goal of ART (defined as achieving undetectable levels of virus by available viral load techniques). With earlier, less potent regimens, many patients were unable to achieve undetectable levels of virus.83–85 In such circumstances, the ability to achieve undetectable levels of virus was more difficult among patients with higher baseline viral load levels and heavier treatment experience.86–92 However, with more potent and tolerable regimens available today, over 80–85% of patients are able to achieve <50 c/mL with their initial regimens and increasingly the <50 c/mL target can be achieved even among very heavily treatment experienced patients.93–95
Initiation of Therapy
The ultimate success of ART is total eradication, or cure, as discussed above. In the case of healthcare workers who have been exposed to infected blood through percutaneous needlestick exposure, the use of ART appears to help abort early infection and, in this regard, is the best example of eradication occurring as a result of ART.96,97 Similarly, prevention of perinatal infection through the use of prepartum and peripartum treatment of the mother and postpartum treatment of the neonate is another example where early infection is either prevented or aborted after exposure through the use of ART.98 Other than these two situations, success with ART in total eradication or cure has not been achieved. Rather, antiretroviral ‘success’ is defined more as the absence of ARV failure rather than as success per se. Therefore, prevention of ARV failure becomes the principal goal of ART.
With the development of a large number of more potent ARV agents, there has been a paradigm shift in the approach to therapy: the focus of therapy has changed from keeping patients alive from this year to the next to keeping patients alive from this decade to the next. The goals of chronic administration of ART can be seen as twofold: to prevent clinical progression and to prevent or delay development of resistance. Over the last decade the guidelines for treatment of HIV infection have vacillated, owing in large part to the emerging realization that a cure was not readily achievable with ART alone.99 The ‘treat early, treat hard’ approach to therapy was initially linked with the idea that complete, maintained viral suppression could result in the eradication of HIV from the body within a short time.61 In addition, confusion regarding the goals of therapy at different stages of disease contributes to the apparent ‘change’ in guidelines. To prevent the emergence of viral resistance, relatively complete viral suppression is required. Therefore, in all newly treated patients the principal goal of treatment should be to achieve virologic suppression to undetectable levels.1,100 While clinical benefit can still be realized with less than maximal suppression of viral load,83,89,101,102 use of more modern drugs has led to the emergence of a new treatment paradigm whereby the target of ARV therapy ideally should be <50 c/mL even in heavily experienced patients.1,4 Yet, for those patients who cannot achieve this degree of virologic suppression, a reduction in HIV RNA levels of 0.5 log10 copies/mL below baseline is associated with relative maintenance of the CD4+ T-lymphocyte count over time, provided this degree of viral suppression is sustained.83,89,102 Therefore for patients who have experienced multiple regimens and cannot achieve <50 c/mL, the goal of therapy changes from prevention of further resistance mutations to prevention of clinical progression. In this setting, a reasonable target is to achieve and sustain viral load values at least 0.5 log10 copies/mL, and preferably 1.0 log10 copies/mL below baseline.
The ‘treat early, treat hard’ approach to therapy was grounded in concepts other than eradication.61,99 Most clinical trials confirm that the first treatment represents the ‘best shot’ at achieving profound suppression of viral replication. Based on current knowledge of HIV pathogenesis, early and profound suppression also prevents the development of resistance by limiting replication, preserving immune system integrity (before there is loss of critical clones of responsive cells), and creating a higher virologic hurdle for emergence of viral resistance.103–105 Although this rationale is clearly sound, the approach rests on assumptions concerning adherence, toxicity, pharmacokinetics/pharmacodynamics, and the absence of meaningful immune system recovery. First, complete adherence to complex ARV regimens is difficult for most patients to maintain.34,106,107 Multiple studies demonstrate a striking relation between adherence and success of ARV therapeutic regimens. Second, although serious toxicity is relatively uncommon with initial treatment for early disease, prolonged exposure to treatment is associated with a number of metabolic and hepatic complications.20,21,108–114 Third, drug pharmacokinetics and pharmacodynamics are subject to variability that can reduce the effectiveness of treatment.31,115 Not all patients are able to achieve and sustain similar levels of intracellular concentrations of drug or are able to metabolize drugs in a predictable fashion.32,116,117 Fourth, although treatment that is initiated late in the course of disease (e.g., at CD4+ T-lymphocyte counts of less than 100 cells/μL) is associated with a worse outcome, treatment started at moderate CD4+ T-lymphocyte counts (e.g., 350 cells/μL) can be accompanied by preservation of immune competence that does not seem to be clinically distinguishable from that seen when starting therapy at earlier stages (e.g., CD4+ T-lymphocyte count of 600 cells/μL).87,118,119
There are additional considerations that argue against universal application of very early treatment.120 Much of the data on progression of HIV disease from the Multicenter AIDS Cohort Study (MACS), which has been used to support earlier intervention, is derived from untreated individuals (Fig. 4-7A).56 The use of data from untreated cohorts, although providing information on the natural history of disease prior to the influence of ART, is anachronistic during the treatment era. Because the impact of treatment on the viral load determinants of progression is immediate, its predictive value is negated once potent treatment is initiated. Moreover, most clinicians do not idly observe progression of disease in their patients without introducing some intervention prior to the onset of profound CD4+ T-lymphocyte depletion or the development of clinical disease. More recent data sets derived from clinical cohorts of treated individuals are more valuable for the ‘treatment era’ (Figs 4-7B and 4-8).119 Data from the ART-Cohort Collaborative, a compilation of treatment experience of over 23 000 patients from over 50 clinical sites, demonstrate little short-term disease progression among patients who initiated therapy with more moderate CD4+ T-lymphocyte counts (e.g., 350–500 cells/μL), even if their viral load values at the time of treatment initiation are high (e.g., >100 000 copies/mL).87,118
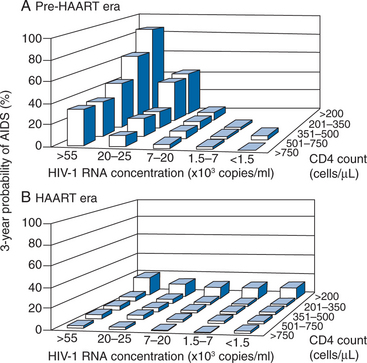
Figure 4-7 Likelihood of progression to AIDS or death in the pretreatment (A) and treatment (B) era.
Used from Egger M, May M, Chene G, et al. Prognosis of HIV-1 infected drug naive patients starting potent antiretroviral therapy: a collaborative analysis of prospective studies. Lancet 360:119–29, 2002, as presented in US Department of Health and Human Services Panel on Clinical Practices for Treatment of HIV Infection. Guidelines for the Use of Antiretroviral Agents in HIV-1 Infected Adults and Adolescents. Washington, DC, DHHS. Available: http://aidsinfo.nih.gov/Guidelines/GuidelinesDetail.aspx?MenuItem=Guidelines&Search=Off&GuidelineID=7&ClassID=1 10 Oct 2006.
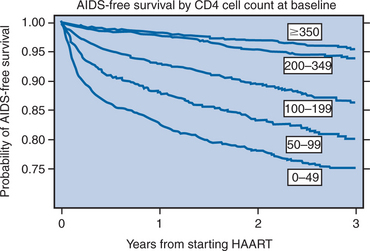
Figure 4-8 Progression of HIV disease to AIDS or death by initial CD4 count.
Used from Egger M, May M, Chene G, et al. Prognosis of HIV-1 infected drug naive patients starting potent antiretroviral therapy: a collaborative analysis of prospective studies. Lancet 360:119–29, 2002.
Based on such considerations, along with the increased frequency of long-term adverse events of treatment, a more conservative approach than ‘treat early, treat hard’ is advocated currently, although as therapies become better tolerated the recommendations continue to be in flux (Table 4-2).1,4 Among asymptomatic patients, treatment may be initiated relatively early (e.g., at CD4+ T-lymphocyte counts of 350–500 cells/μL; while guidelines suggest consideration of treatment at these CD4+ T-lymphocyte counts, some experts now recommend treatment at this level) rather than very early (e.g., CD4 counts >500 cells/μL) using ARV combinations that are likely to reduce the viral load below 50 copies/mL.1,4,5 In contrast, all symptomatic patients should have treatment initiated regardless of CD4+ count and viral load. Similarly, all HIV-infected pregnant women should have treatment initiated by the beginning of the second trimester, regardless of viral load or CD4+ count, to eliminate the transmission of HIV to her baby. Once the mother has delivered, therapy can be stopped among those women who had initial CD4+ counts above 350–500 cells/μL and who are not breast-feeding (Table 4-2).1,4
Table 4-2 Recommendations for Initiating ART in Treatment-Naive Adultsa with Chronic HIV Infection
| Parameter | Recommendation: IAS-USA1 | Recommendation: HHS4 |
|---|---|---|
| Symptomatic HIV Disease | ART is recommended | ART is recommended |
| Asymptomatic HIV Disease | ||
| CD4+ ≤200 cells/μL | ART is recommended | ART is recommended |
| CD4+ <350 cells/μL but >200 cells/μL | ART should be consideredb and the decision is individualized (see text); monitoring and counseling for HIV transmission prevention should continue if therapy is deferred | Treatment should be offered following discussion of pros and cons of therapy |
| CD4+ >350 cells/μL | ART is generally not recommendedc; monitoring and counseling for HIV transmission prevention should continue | VL >100 000 c/mL, most clinicians recommend deferring ART, but some would treat; VL < 100 000 c/mL, defer therapy |
| but ≤500 cells/μL | ||
| CD4+ >500 cells/μL | ART is generally not recommended; monitoring and counseling for HIV transmission prevention should continue | VL > 100 000 c/mL, most clinicians recommend deferring ART, but some would treat; VL < 100 000 c/mL, defer therapy |
b The closer the CD4+ cell count is to 200/μL, the stronger the recommendation, particularly if the plasma viral load is high (>100 000 copies/mL) or if the CD4+ count is declining rapidly (>100 cells μL−1 year−1).
c Consider treatment for patients with high plasma viral load or with rapid decline of CD4+ cell count.
Adapted from IAS-USA Guidelines 20061
Drugs cannot work if they are not taken. Creating a regimen that is relatively easy for the patient to take therefore is essential when designing initial treatment (Fig. 4-9A). Moreover, frequently missed doses not only decrease the ability of the drugs to work, it is a recipe for the development of ARV resistance. A study comparing outcomes among prisoners versus patients who participated in clinical trials using the identical regimens from the clinical trials, demonstrated that the prisoners, who had drugs administered under conditions of directly observed therapy, had substantially better virologic outcomes (90% vs 75% achieved viral load values of less than 400 copies/mL).121 These data underscore the importance of drug-taking behavior, commitment to the regimen, simplicity of regimens, and overall adherence to the success of the initial regimen.
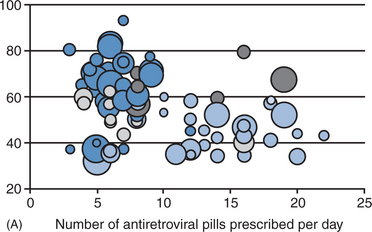
The potency of the initial regimen is critical to long-term success. Regimens for initial treatment should be of sufficient potency to ensure a high likelihood of achieving viral load levels below the limit of detection (<50 c/μL). Preexisting drug resistant virus, which has been reported with increased frequency (between <3% and 14%), significantly hampers the potency of the initial regimen. Current guidelines suggest obtaining a pre-ARV treatment genotypic resistance test for any patient living in an area where a high prevalence (>5%) of resistant virus is known to exist, for patients presenting with acute seroconversion syndrome, and for pregnant women in selected cases.
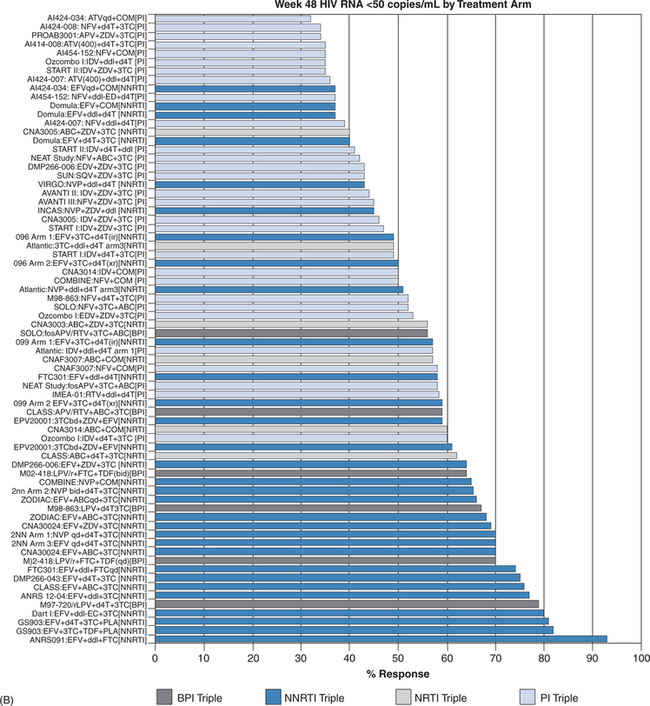
Once a decision has been made to initiate therapy the proper choice of regimen is essential. Meta-analyses have shown that efavirenz plus two nucleoside reverse transcriptase inhibitors (NRTIs) demonstrates ARV effectiveness activity comparable, if not superior, to that of ritonavir boosted protease inhibitor (PI)-containing regimens (Fig. 4-9B).30,122,127 However, ritonavir-boosted regimens are more likely to be associated with hypertriglyceridemia and possibly insulin resistance and lipodystrophy, reinforcing the trend for use of PI-sparing regimens as initial therapy. Unboosted PI-containing regimens are universally inferior to boosted-PI and most non-nucleoside reverse transcriptase inhibitor (NNRTI) containing regimens and should be used only in selected clinical situations, such as in some active intravenous drug users, in those with significant hepatic compromise, or in patients requiring therapy for concomitant diseases where the drugs interact with ritonavir or NNRTI agents. In head-to-head studies, efavirenz has generally been more effective (by intent to treat) than nevirapine, although the outcomes were at the margin of statistical significance. Nevirapine should not be used in patients with higher CD4+ T-lymphocyte counts (>250 cells/μL for women; >350 cells/μL for men) owing to a higher risk of hepatic toxicity.27 Efavirenz should generally be avoided in pregnant women, especially in the first trimester when the neural tube maturation is occurring. The preferred and alternative regimens according to the HHS guidelines are shown in Table 4-3.
Table 4-3 Initial Treatment (HHS Guidelines+)
| Preferred Option | |
|---|---|
| NNRTI Option | NRTI Options |
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |
