Chapter 8 Stavudine
STRUCTURE
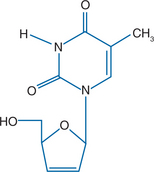
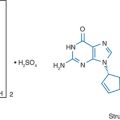
Stavudine (2′,3′didehydro-2′,3′-didoxythymidine: d4T), like zidovudine, is an analog of thymidine (Fig. 8-1). It differs from thymidine by replacement of the hydroxyl group at the 3′ position by a hydrogen. It is relatively water soluble with solubility of 83 mg/mL in water at 23°C.1–3
MECHANISM OF ACTION AND IN VITRO ACTIVITY
Like other nucleoside analogs, stavudine is not active as the parent compound, but must be phosphorylated to the active intracellular form, stavudine triphosphate.1,4 The parent compound enters the cell by passive diffusion.5 It is phosphorylated by thymidine kinase, thymidylate kinase, and pyrimidine diphosphate kinase.6 The initial phosphorylation by thymidine kinase appears to be the rate-limiting step in the activation of stavudine.7 Unlike zidovudine, there is no accumulation of the monophosphate. The mono-, di-, and triphosphate forms of d4T are present in approximately 1:1:1 ratio, and increasing the extracellular concentration of stavudine results in proportional increases in intracellular concentration of the active form.6–8 The accumulation of zidovudine monophosphate and the difficulty in achieving high levels of the active triphosphate may limit the antiviral activity of zidovudine and contribute to toxicity.9,10 In contrast to zidovudine, stavudine does not result in depletion of the intracellular pools of thymidine-5′-triphosphate in bone marrow progenitor cells. This was hypothesized to explain the lack of hematologic toxicity,11 but other studies have suggested that depletion of thymidine-5′-triphosphate does not explain zidovudine’s bone marrow toxicity.12,13
The triphosphate form of stavudine acts as a competitive inhibitor of reverse transcriptase by competing with the natural substrate, 2′-deoxy-thymidine-5′-triphosphate.1,6,14,15 The alterations in the ribose ring at the 3′ position prevent formation of a new 3′-5′ phosphodiester bond with the next nucleotide. Therefore, stavudine also acts as an obligate chain terminator.
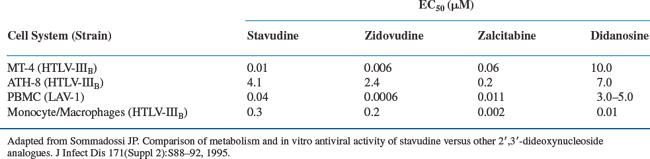
Stavudine shows antiviral activity in vitro against both HIV-1 and HIV-2, but not against hepatitis B, herpes simplex, or CMV.8,16,17 The 50% inhibitory concentration (IC50) for stavudine against laboratory and clinical isolates ranges from 0.009 to 4.1 μM (0.002–0.9 μg/mL) depending on the cell type and the assay system (Table 8-1).1,8,11,13 Like zidovudine, stavudine is primarily active in HIV-infected activated cells, such as phytohemagglutinin-stimulated peripheral blood mononuclear cells, probably because thymidine kinase is an S-phase-specific enzyme.13,18,19 Stavudine exhibits partial cross-resistance with zidovudine; isolates with multiple zidovudine resistance mutations (M41L, D67N, T215Y) have reduced susceptibility.20–23 Stavudine shows additive or synergistic activity in vitro when combined with didanosine, lamivudine, nevirapine, saquinavir, indinavir, and nelfinavir.24–27 Zidovudine and stavudine compete for activation by thymidine kinase.28 When they are co-administered in vivo, the amounts of stavudine triphosphate are reduced relative to zidovudine triphosphate.6 As might be predicted, the combination of zidovudine and stavudine shows antagonism in vitro against zidovudine resistant strains of HIV.24
PHARMACOKINETICS
Stavudine is well absorbed. Initial studies suggested a bioavailablity of 82–86%,2,29,30 but population pharmacokinetic analysis on 33 patients calculated bioavailability of greater than 99%31,32, consistent with complete bioavailability observed in mice.33 The area under the time-concentration curve (AUC) is identical when stavudine is taken while fasting or with a high fat meal, but the maximal concentration (Cmax) is reduced when given with a meal.34 Peak plasma concentrations increase in a dose-related manner. After a 0.67 mg/kg dose, the peak serum concentration of 1.2 μg/mL is reached in within 1 h.29 The estimated volume of distribution is 0.53 L/kg,29,33 and there is negligible protein binding.
Plasma concentrations of stavudine decline in a biphasic manner, independent of dose. The mean plasma half-life following single oral doses in HIV-infected volunteers was 1.6 ± 0.8 h. The intracellular half-life, however, is between3 and 4 h.19 Interpatient variation in the absorption and excretion of stavudine is relatively low.35 About 40% of stavudine is excreted unchanged in the urine; therefore, dose adjustment is necessary for patients with renal failure (creatinine clearance <50–mL/min–see recommendations for use).36 Two small studies have examined the clearance of stavudine in patients on hemodialysis (Bristol-Myers Squibb Investigational Drug Service written communication, Nov 1996). Cmax was unchanged but the terminal half-life was prolonged up to 12 h. The mean hemodialysis clearance value of stavudine was 120 ± 18 mL/min. No dose adjustment is necessary in patients with hepatic impairment.37
Based on primate studies, the nonrenal metabolism is thought to involve cleavage of the sugar to yield thymine, which is degraded to beta-aminoisobutyric acid, or used in the pyrimidine salvage pathway.38
Stavudine crosses the blood–brain barrier. Levels above the IC50 were measured in brain tissue of rats, dogs, and monkeys after a single oral dose of 25 mg/kg.33 Single-dose studies using a 40 mg oral dose in healthy volunteers demonstrated a mean concentration of 61 ng/mL at 4–5 h, with a mean CSF to plasma ratio of 40% (Bristol-Myers Squibb Investigational Drug Service, written communication, Apr 1997). Foudraine reported CSF concentrations of stavudine at week 12 in 17 HIV-infected patients treated with stavudine and lamivudine. The concentration of stavudine ranged from 0.2 to 0.27 μmol/Land the mean CSF to plasma ratio was 38%.39 The concentration exceeded the mean IC50 for clinical isolates. Foudraine and others40 also made the important observation that calculation of the CSF to plasma ratio can be misleading, since the ratio is highly dependent of the timing of sampling. This is because the clearance rate from plasma is much faster than from CSF. Absolute concentrations of drug relative to antiviral activity may be more reliable indicators. In HIV-infected children on stable dosing, CSF stavudine concentrations, obtained ∼2–3 h after oral doses, ranged from 16% to 97% of simultaneous plasma concentrations.41 The clinical relevance of stavudine concentrations in the CSF was suggested in Prometheus, a trial that randomized patients to ritonavir and saquinavir with or without stavudine. Twelve of 13 patients on the stavudine arm suppressed CSF HIV RNA to undetectable levels compared to only four of 14 of those not on stavudine.42
In pregnant macaques, stavudine reached the fetal circulation with a maternal to fetal ratio of 77–81%.43 In ex vivo studies using human placenta, stavudine crossed the placenta via rapid, nonfacilitated, nonsaturable diffusion, with similar pharmacokinetic properties to zidovudine.44 Concentrations within placental tissue were approximately twofold lower for stavudine than for zidovudine, however, probably reflecting stavudine’s lower lipid solubility.44
TOXICITY
Stavudine is generally well tolerated in the short term and side effects are modest. Thus, it was initially thought of as one of the safest nucleosides. However, it required several years of widespread clinical use before the long-term toxicities were better understood. These long-term toxicities are the most clinically important, including mitochondrial toxicity, lipoatrophy, lactic acidemia and lactic acidosis (discussed in more detail in Chapters 73 and 74). These long-term adverse events now limit the long-term use of stavudine to exceptional clinical situations, such as in patients who harbor HIV with a K6 mutation and cannot tolerate zidovudine, and in resource-constrained countries where other nucleoside agents are not available.
Preclinical studies predicted the major clinical toxicities of stavudine. In vitro, stavudine is substantially less toxic to bone marrow precursors than zidovudine.45,46 However, stavudine inhibits outgrowth of neurites in an in vitro model of neurotoxicity, with slightly less toxicity than didanosine or zalcitabine, but substantially more than zidovudine.47,48 The neurotoxicity of these nucleoside analogs was thought by some investigators to be due to inhibition of mitochondrial DNA polymerase-γ.49
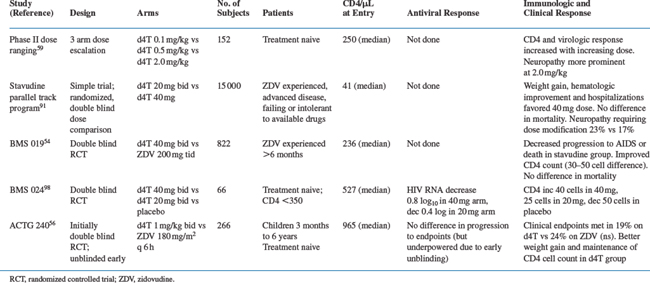
Clinical toxicities of stavudine monotherapy from phase III studies are shown in Table 8-2. Stavudine does not cause significant bone marrow suppression.50–54 Initial phase I and phase II dose-ranging trials demonstrated that peripheral neuropathy, and to a lesser extent, elevations of hepatic transaminases are the major dose-limiting toxicities of stavudine. The maximum tolerated dose of stavudine established in phase I studies was 2 mg kg−1 day−1.50–53
Table 8-2 Clinical Toxicities of Stavudine from a Phase III Randomized Clinical Trial of Monotherapya
| Rate per 100 person years | ||
|---|---|---|
| Adverse Event | Stavudine (40 mg bid) n = 417 | Zidovudine (200 mg tid) n = 405 |
| Headache | 36 | 38 |
| Chills/fever | 34 | 40 |
| Diarrhea | 33 | 34 |
| Rash | 27 | 27 |
| Nausea and vomiting | 26 | 35 |
| Abdominal pain | 23 | 21 |
| Myalgia | 21 | 27 |
| Insomnia | 20 | 24 |
| Pancreatitis | <1 | <1 |
| Neuropathy any grade | 11.7 | 3.9 |
| Neuropathy grade 3–4 | 2 | 1 |
| Hyperamylasemia (>1.4 X ULN) | 12.6 | 10.7 |
| Elevated AST any grade | 65.6 | 44 |
| Elevated AST >5 X ULN | 8.6 | 9.3 |
| Elevated ALT any grade | 69.1 | 41.3 |
| Elevated ALT >5 X ULN | 12.1 | 9.9 |
| Anemia grade 3–4 | 0.6 | 14.2 |
| Neutropenia grade 3–4 | 3.0 | 6.4 |
a Events among patients on zidovudine are included for comparison.
From Spruance SL, Pavia AT, Mellors JW, et al. Clinical efficacy of monotherapy with stavudine compared with zidovudine in HIV-infected, zidovudine-experienced patients. A randomized, double-blind, controlled trial. Bristol-Myers Squibb Stavudine/019 Study Group. Ann Intern Med 126:355–63, 1997 and Final Report of Protocol AI455-019.
The risk of peripheral neuropathy is dose related. In an analysis of data from three phase 1 studies and one phase IIstudy, the incidence of neuropathy (of any severity) was 17–21/100 person years in patients receiving doses of 0.5 mg kg−1 day−1, 21/100 person years at 1.0 mg kg−1 day−1, and 41–66/100 person years at 2.0 mg kg−1 day−1. Patients receiving 4 mg kg−1 day−1 for extended periods had a cumulative incidence of peripheral neuropathy of 64–71%.53 In BMS 019, the phase III efficacy trial among zidovudine-experienced patients (using the currently recommended dose of 40 mg po bid, ∼1 mg kg−1 day−1), the rate of neuropathy sufficient to require dose modification was 11.7/100 person years among stavudine-treated patients compared to 3.9/100 person years in the zidovudine group.54 The occurrence of neuropathy was strongly associated with a baseline diagnosis of AIDS, preexisting neuropathy, and with low CD4 counts at study entry.
Neuropathy associated with stavudine is usually reversible when the drug is discontinued or the dose is modified promptly. Among 56 stavudine-treated patients who developed neuropathy in the phase III trial, 63% had complete resolution of neurologic symptoms within a median of 17 days. These patients all tolerated rechallenge at half of the original dose.54 In pediatric trials, peripheral neuropathy has been rare.41,55–57 Higher rates of neuropathy have been observed in patients treated with stavudine in combination with didanosine and hydroxyurea.58
Modest elevations of hepatic transaminases are common with stavudine therapy, but significant elevations and clinical hepatitis are uncommon. Grade III or IV elevations of hepatic transaminases (greater than five times the upper limit of normal) occurred in 9–13% of patients receiving doses of 2.0 mg kg−1 day−1 of stavudine in the phase I and II studies.53,59 Significant elevations were not apparently related to dose, but were significantly associated with abnormal baseline levels. In the phase III study, grade III or IV elevations of alaninine aminotransferase (ALT) or aspartate aminotransferase occurred in similar rates in the stavudine and zidovudine groups (12/100 person years vs 10/100 person years) but milder elevations were common and significantly more frequent in the stavudine group (69 vs 44/100 person years).54
Lactic acidemia, lactic acidosis, and hepatic steatosis were recognized early as rare but severe complications of nucleoside analogs, including zidovudine, zalcitabine, didanosine, and stavudine.60–64 Severity ranges from asymptomatic lactic acidemia to fatal fulminant disease. Symptoms are nonspecific and include fatigue, nausea, vomiting, and abdominal pain. Mitochondrial dysfunction appears to be an underlying defect,65–70 although other factors, including the rate of lactate clearance, may be critical.71,72 A number of case reports and case series have implicated stavudine in lactic acidemia and lactic acidosis.62,70,73–78 However, it has been difficult to determine the risk associated with stavudine relative to other nucleoside analogs, and to determine the role of host factors.79 However, there appears to be a hierarchy of risk with ddC > didanosine > stavudine > zidovudine  tenofovir, abacavir, or lamivudine.
tenofovir, abacavir, or lamivudine.
A study that involved 18 months of prospective monitoring of 349 patients found lactate levels to rise modestly after the initiation of stavudine- or zidovudine-based HAART.78 However, the increase was an average 0.23 mmol/L greater in those on stavudine. Severe hyperlactatemia and hepatic steatosis, however, were rare. In Africa, where stavudine-based combination therapy is widely used, symptomatic lactic acidosis is being increasingly recognized, although the incidence and risk factors remain to be defined.80 The combination of stavudine and didanosine is associated with markedly increased risk of lactic acidosis, and hepatic steatosis.
It has been proposed that stavudine-induced mitochondrial dysfunction plays a role in lipodystrophy, especially lipoatrophy.81–83 (See also Chapters 72 and 73.) Many studies are plagued by the complexity of overlapping syndromes, cross-sectional design, and multiple confounders and interactions, both biologic and statistical. Nucleoside analog use, duration of therapy, age, and gender have been consistent risk factors for lipoatrophy.84,85 Many studies identified an association between stavudine and lipodystrophy although early studies were not able to adequately control for common prescribing of stavudine or duration of therapy.82,86 However, prospective cohort studies do show an increased risk of lipodystrophy associated with stavudine use. Among 277 patients participating in the Western Australia cohort, stavudine-containing regimens were associated with an increased relative risk of fat wasting, as were PI use, white race, age, and duration of nucleoside analog therapy.85 Among 1035 patients participating in the British Columbia drug treatment program, lipo-dystrophy was diagnosed based on prospectively collected self-report. Lipodystrophy syndrome was independently associated with age, use of PI therapy, being employed, use of alternative therapy and duration of stavudine therapy87 (see Chapter 73).
Perhaps the clearest demonstration of the role of stavudine comes from several trials which examined the effect of stopping stavudine and changing to other nucleoside analogs on lactic acidosis, mitochondrial function and lipoatrophy. In a study of 118 patients with lipoatrophy who were switched to abacavir (n = 86) or zidovudine (n = 32), lactic acidosis stabilized or improved.88 Modest improvements in limb fat were measured, but not in facial lipoatrophy.89 The high prevalence of facial lipoatrophy and the lack of improvement in facial fat after stopping stavudine has led to a significant decrease in the use of this drug in clinical practice. Improvements in the DNA content of adipocyte mitochondria have also been demonstrated after switching.90
In contrast to didanosine, stavudine alone is not clearly associated with increased risk of pancreatitis or hyperamylasemia.54,91,92
Stavudine is classified as FDA pregnancy category C. Long-term animal carcinogenicity studies are negative.93 Animal toxicology studies of stavudine in pregnancy show no specific embryopathy. Exposure of early embryos to high levels of stavudine (10 μmol/L) inhibited blastocyst formation.94 There was no reduction in fertility at doses greater than 200 times the human levels. Teratology studies using 400 times the human serum concentrations in rats and 183 times the human serum concentration in rabbits did not show any increase in birth defects. In rats exposed to the highest doses, there were decreases in sternal bone calcification and mild increases in neonatal mortality. These changes are consistent with nonspecific toxicity.93,95,96
EFFICACY
Since 1996, combination therapy has become the standard of care. However, in many trials of combination therapy, it is not possible to discern the individual role of the nucleoside analog component. Therefore, it is useful to review data on monotherapy (Table 8-3) and dual-therapy trials (Table 8-4).
Monotherapy
In a phase II dose-ranging study, beneficial effects on CD4 cell count, p24 antigenemia, cellular viremia, and weight gain were observed at doses of 0.5 mg kg−1 day−1 or 2.0 mg kg−1 day−1.59 Little or no response was observed at 0.1 mg kg−1 day−1. The median CD4 cell increase was 20–30 cells/mm3; greater responses were observed in zidovudine-naive patients. The greatest antiviral response was seen at 2.0 mg kg−1 day−1, with a 77% decrease in the median titer of infected PBMCs. Another small study of 15 patients demonstrated median drops in the infectious titer of 1–2 log10 and a median decrease in viral RNA of 0.5 log10 at 52 weeks.97 Katlama and colleagues examined short-term antiviral effect of stavudine among 66 treatment-naive subjects randomized to either stavudine at 20 mg bid (∼0.5 mg kg−1 day−1), stavudine 40 mg bid (1.0 mg kg−1 day−1) or placebo.98 Responses tended to favor the 40 mg bid dose compared to the 20 mg bid dose. The median CD4 increase at week 8 was 63 cells/mm3 in the 40 mg bid group against 33 cells in the low-dose group and a 50-cell decline in the placebo group. The median decrease in cellular viremia was 1.0 log10 versus 0.7 log10, and the median decrease in plasma HIV RNA at week 12 (measured by NASBA) was 0.8 log10 versus 0.4 log10. In ACTG 306, 34 treatment-naive patients were randomized to stavudine monotherapy at 40 mg bid. At 24 weeks, the mean decrease in viral load was 0.55 log10, or 1.04 after adjustment for censoring.99 In patients with prolonged zidovudine therapy, the response to stavudine is attenuated. Among patients with very prolonged zidovudine monotherapy (mean 3.6 years) in ACTG trial 175 who were switched to stavudine monotherapy, the virologic response at 48 weeks was modest (mean decrease 0.18 log10).100
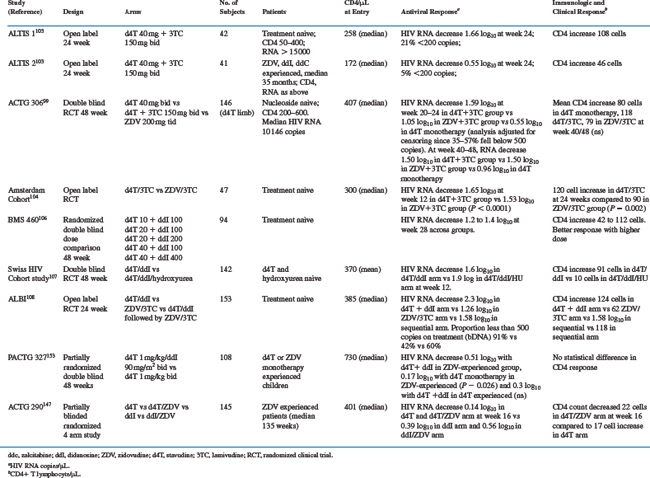
The only clinical endpoint study of stavudine monotherapy (BMS 019) compared stavudine (40 mg bid) with continued zidovudine in 822 patients with CD4 cell counts of 50–500 at least 6 months of prior zidovudine treatment.54 Median duration of follow-up was 115 weeks. Patients receiving stavudine reached clinical endpoints (AIDS-defining events or death) at a significantly lower rate than those randomized to continue zidovudine (26/100 person years vs 32/100 person years; relative risk 0.75; P = 0.03; Fig. 8-2). This benefit was apparent in all strata of baseline CD4 cell count. Survival was not significantly different, but there was a trend favoring longer survival in the stavudine group (relative risk 0.74; P = 0.066). Patients assigned to stavudine remained on initial therapy significantly longer (79 weeks compared to 53 weeks). Quality of life as measured by the MOS SF-36 was significantly better among stavudine-treated patients at week 12, but by week 36 the difference was not significant.101 Patients in the stavudine group had significantly less anemia and neutropenia, better weight gain. The CD4 count initially increased a median of 20 cells, but then declined. The count remained 30–40 cells/mm3 higher than the zidovudine group (Fig. 8-2). Viral RNA was not measured in this study.

Stavudine Combined with Lamivudine
Because of the convenient dosing and the favorable side effect profile, the combination of stavudine and lamivudine became popular before clinical trial data were available to demonstrate efficacy.102 In vitro, the combination shows additive to synergistic effects.24 The combination appears at least as active as zidovudine and lamivudine.
In an open-label prospective study (ALTIS I) among 42 treatment-naive patients,103 the combination of stavudine and lamivudine resulted in a median decrease in viral RNA of 1.96 log10 at 4 weeks, sustained at −1.66 log10 at 24 weeks with an increase in the CD4 count of 108 cells/mm3 at 24 weeks. RNA levels were below 500 copies/mL at week 24 in 30%. In a parallel study of 41 antiretroviral-experienced but stavudine- and lamivudine-naive patients (ALTIS II), more modest effects were observed. The median maximum change in HIV RNA was −1.3 log10 at week 4, but only −0.55 log10 at week 24 accompanied by a CD4 increase of 46 cells/mm3.
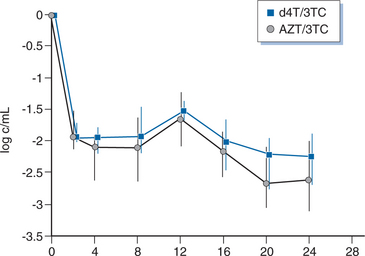
Two studies have compared stavudine plus lamivudine with zidovudine plus lamivudine. ACTG 306 was a six-arm study among antiretroviral-naive patients.99 The stavudine arm compared stavudine monotherapy, stavudine plus lamivudine, and zidovudine plus lamivudine. Adjusted for censoring, the change in viral load at week 20–24 was −0.55 log10 in the stavudine monotherapy arm, −1.59 log10 in the stavudine/lamivudine arm, and −1.05 log10 in the zidovudine arm. The difference favoring stavudine/lamivudine over zidovudine was of borderline statistical significance (P = 0.052) at week 24, but no difference was seen at week 48. A low baseline viral load complicates the analysis. Using an ultrasensitive assay (lower limit 20 copies/mL), there was no difference between stavudine/lamivudine and zidovudine/lamivudine (−1.64 vs −1.57 log10). In another randomized trial among 47 patients who received stavudine/lamivudine or zidovudine/lamivudine for 12 weeks, the viral load decreased by −1.65 compared to −1.53 log10 (Fig. 8-3). However, all patients had developed lamivudine resistance by 12 weeks.104 In 28 patients who underwent lumbar puncture before and after therapy, both regimens resulted in CSF HIV RNA becoming undetectable by quantitative PCR in all patients.39



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


