Spinal Canal
Tumors of the spinal cord and cauda equina account for 3% to 4% of central nervous system (CNS) tumors overall and 6% of CNS tumors in children.1 Spinal canal tumors are classified by the World Health Organization (WHO) according to histological types. Clinically, they are also characterized by their location relative to the protective layers of the spinal cord as either extradural, intradural–extramedullary, or intramedullary (Fig. 37.1). Intramedullary lesions arise from the intrinsic substance of the spinal cord. Histologically, intramedullary spinal cord neoplasms include gliomas such as astrocytoma, ependymoma, and oligodendroglioma. Intradural–extramedullary tumors arise from the connective tissues, blood vessels, or coverings adjacent to the cord or cauda equina. Common histologies include nerve sheath tumor, meningioma, and ependymoma. Extradural tumors are most commonly metastatic, although primary tumors in this compartment may occur as well. Primary extradural tumors arising from the vertebral bodies may include benign tumors such as osteoid osteoma, osteoblastoma, or aneurysmal bone cysts, or as malignant tumors such as plasmocytoma or myeloma, chordoma or chondrosarcoma, osteosarcoma, and Ewing’s sarcoma. Other primary extradural tumors arising outside of the vertebral body include epidural hemangiomas, lipomas, extradural meningiomas, nerve sheath tumors, and lymphomas.
Radiation therapy is an important modality in the management of both primary and metastatic tumors involving the spinal canal. This chapter focuses primarily on the management of primary spinal cord tumors. Primary extradural tumors are usually managed in a manner similar to histopathologically identical tumors arising at other locations and will not be discussed here in detail. The management of metastatic tumors involving the spinal canal is discussed in Chapter 93.
FIGURE 37.1. Neoplasms affecting the spinal cord. A: Normal transverse spine. The spinal cord is enveloped by the pia, arachnoid, and dura mater, which are housed in the spinal canal and surrounded by ligaments supporting the vertebral bony structures. The subarachnoid space contains cerebrospinal fluid (brown). B: Transverse spine with extradural mass. An extradural mass (e.g., metastasis) from the vertebral body is compressing the dural sac and the spinal cord from the anterior direction. The subarachnoid space becomes obliterated at that level, causing a myelographic block. C: Transverse spine with an intradural–extramedullary mass. The mass, typically a meningioma or nerve sheath tumor, is compressing the spinal cord and roots in the dural sac, causing a myelographic block with a laterally displaced cord and, at times, producing a capping contour of contrast border. D: Transverse spine with intramedullary mass. An intramedullary mass (astrocytoma or ependymoma) is infiltrating and expanding the spinal cord within the dural sac, causing a myelographic block.
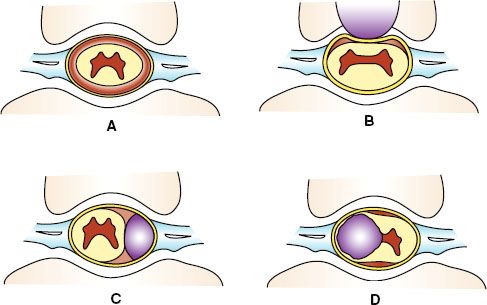
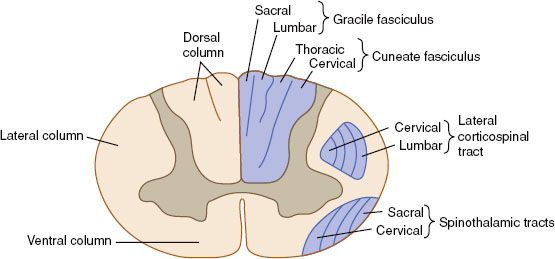
FIGURE 37.2. Somatotopic organization of the cervical spinal cord in transection. (From Waxman S, Clinical neuroanatomy. New York: McGraw-Hill, 2010, with permission.)
 ANATOMY
ANATOMY
Spinal Cord
The spinal cord is a slender cylinder composed of functional segments corresponding to 31 pairs of spinal nerves: 8 cervical, 12 thoracic, 5 lumbar, 5 sacral, and 1 coccygeal. In contrast to the brain, the white matter of the spinal cord is located in the periphery and surrounds the central gray matter. The gray matter contains the cell bodies of sensory, motor, and autonomic neurons. On cross-section, the gray matter is a butterfly-shaped region with anterior horns controlling motor function, lateral horns (in the thoracic and upper lumbar region) controlling autonomic functions, and posterior horns involved in sensation. The white matter contains the axonal elements of neurons that transmit impulses to and from the brain. As in the brain, the axons of the spinal cord white matter possess a myelin sheath formed by the cytoplasmic extension of glial cells. Schwann cells sheath the spinal nerves that enter and exit the spinal cord. The spinal cord is organized into somatotopically distinct regions (Fig. 37.2). The lateral and anterior spinal cord white matter contains the nerve tracts that are involved with fine motor control and tone, including the corticospinal tracts. The spinocerebellar tracts transmit muscle stretch and tone sensation from the extremities to the cerebellum. The lateral spinal thalamic tract is located laterally near the spinal cord surface and carries ascending crossed pain fibers to the thalamus. The dorsal columns transmit fine touch and positional sensation from the extremities to the brain. Because of its serial organization, injury to the spinal cord results in characteristic neurologic findings that depend on the location of the insult.
The spinal cord is surrounded by the meninges, which is composed (from outer to inner) of the dura mater, arachnoid, and pia mater. The pia mater covers the spinal cord and its blood vessels. This layer condenses laterally into approximately 20 pair of dentate ligaments, which suspend the cord to the dura mater. The dura mater forms a dense, fibrous barrier between the bony spinal canal and the spinal cord. The dural sac ends inferiorly at S2-3 but the dura continues with the filum terminale down to the coccyx. The arachnoid mater resides between the dura mater and the pia mater. The arachnoid encloses the subarachnoid space filled with cerebrospinal fluid (CSF). The subarachnoid space follows the arachnoid down to the end of the dural sac.
The growth of the vertebral column during childhood takes place at a rate and extent greater than that of the spinal cord itself. By adulthood, the spinal cord is nearly 25 cm shorter than the vertebral column and ends near the level of the L1 vertebral body. Because of this differential growth, the exit level of each pair of spinal nerves in the spinal cord is usually higher than the corresponding vertebral body level. For example, in adults the C8 nerve root leaves the cord at the C6 vertebral body, the T6 nerve at the T3 vertebral level, and the T12 nerve at the T9 vertebral level. All of the lumbar nerves exit the spinal cord from vertebral levels T10 through T12, and all of the sacral nerves exit the spinal cord near the L1 vertebral level. The lower lumbar, sacral, and coccygeal nerves form the cauda equina, the collection of nerves that fills the thecal sac below L1. At its most caudal extent, the cord tapers to a thin segment, the conus medullaris. It is tethered to the coccyx by the filum terminale, a dense thread of pia mater.
Spinal Canal
The posterior body surfaces and neural arches of the vertebrae form the vertebral foramina, which in continuity form the spinal canal. Vertebral foramina are triangular in the lumbar and cervical regions, where the cord is mostly mobile, and round in the thoracic region. The spinal canal is lined with ligaments, including the posterior longitudinal ligament on its anterior wall, the flaval ligaments between adjacent arches, and the interspinous ligaments between the spinous processes. At each vertebral level, a spinous process protrudes from the posterior aspect of the neural arch, and transverse processes extend from the lateral edges of each arch. The laminae are those portions of the neural arch between the spinous and transverse processes, and the pedicles lie between the transverse processes and the body. At the intersection between the laminae and pedicles are superior and inferior paired articular facets. The superior articular facets are synovial joints that articulate with the inferior articular facets of the vertebra immediately above. The paired pedicles of each vertebra are notched at their superior and inferior edges such that the notches from two contiguous vertebra form an intervertebral foramen, through which the spinal nerve courses.
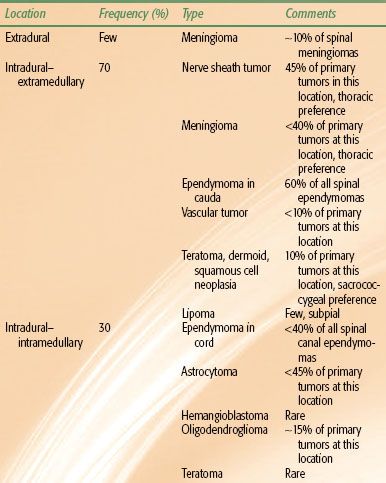
TABLE 37.1 PRIMARY SPINAL CANAL TUMORS: LOCATIONS, TYPES, AND FREQUENCIES
 EPIDEMIOLOGY
EPIDEMIOLOGY
Primary spinal canal tumors comprise 3% to 4% of all primary CNS tumors.1 Their incidences vary by location and age. Primary spinal canal tumors appear to be more common in non-Hispanic whites than Hispanics or non-Hispanic blacks.2 Although the incidence of spinal cord ependymoma has increased significantly over the past 30 years, the incidence of other glioma subtypes has remained stable.3
In adults, nearly two-thirds of all intradural tumors are extramedullary and are typically nerve sheath tumors, meningiomas, or ependymomas. The other third of intradural tumors are intramedullary, with the most common histologies being astrocytoma and ependymoma, followed by hemangioblastoma and other tumor types (Table 37.1). Overall, extramedullary nerve sheath tumors and meningiomas represent the most common spinal canal neoplasms, followed by intramedullary ependymomas and astrocytomas.
Primary tumors of the spinal canal are relatively more frequent in children, accounting for 6% of pediatric CNS tumors.1 More than 50% of pediatric patients are younger than 10 years of age.4,5 In a review of 872 children with intraspinal tumors, 36% had intramedullary tumors, 27% had intradural extramedullary tumors, and 24% had extradural tumors (13% were unclassified).5 Nearly 75% of pediatric intramedullary tumors were astrocytomas or gangliogliomas and a few were ependymomas. Approximately 25% of the intradural–extramedullary tumors were ependymomas, followed in incidence by dermoids (23%), teratomas (16%), nerve sheath tumors (14%), lipomas (13%), and meningiomas (9%).6
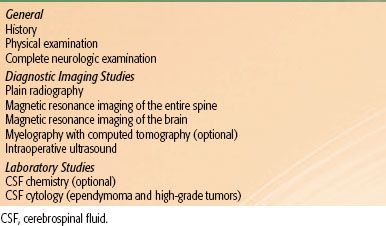
TABLE 37.2 DIAGNOSTIC WORKUP FOR PRIMARY SPINAL CORD TUMORS
 NATURAL HISTORY
NATURAL HISTORY
Most primary tumors of the spinal canal are histologically benign. Despite this, they are often the cause of significant disability because they compress or invade the spinal cord and interfere with neurologic function. Intramedullary tumors produce neurologic damage by local invasion or cystic compression of the cord, whereas extramedullary lesions compress, stretch, or distort the cord or the spinal nerves. Primary spinal cord tumors may be focal or relatively localized in some patients but may involve nearly the entire length of the cord in others. In one report, 73% of affected children presented with widening of the entire spinal cord from the medulla or cervical medullary junction to the conus medullaris.7 These “holocord” tumors typically consist of a discrete solid mass and an associated cystic component or syrinx that extends over a significant length of the spinal cord. Local tumor progression is the dominant form of treatment failure of spinal cord tumors. CSF seeding is possible but uncommon.8–10,11 Because the CNS has no lymphatics, spread to lymph nodes is not seen with spinal canal tumors. Extraneural spread is rare, occurring with an overall incidence of 0.96% in a cohort of 28,441 CNS cancers identified in a Surveillance, Epidemiology, and End Results (SEER) analysis. Interestingly, cancers of the spinal cord had a higher risk of extraneural spread relative to cerebral tumors.12 The major causes of death in patients with spinal canal tumors are complications of paraplegia or quadriplegia such as infection or respiratory compromise.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Pain is the most common presenting symptom, affecting approximately 72% of patients.13 Often the pain is localized to the region of involvement and may be present for a long time before the patient manifests localizing neurologic signs. Radicular pain, a result of pressure on nerve roots, reflects the distribution of the involved root and indicates that conduction is intact. Numbness replacing pain is a more advanced sign that indicates compromise of spinal nerve or nerve tract conduction. Extramedullary tumors can cause distention of the dura with severe pain in the region of the tumor that is characteristically aggravated by recumbency because of venous congestion. Thus, pain is often worse at night.7 Movement or the Valsalva maneuver also may worsen pain. Less commonly, pain is characterized as a burning sensation in one or more extremities.
Other symptoms of CNS involvement include weakness (55% of patients), sensory deficits (39%), and sphincter dysfunction (15%).13 Low-grade tumors generally have a more prolonged duration of symptoms than high-grade tumors. Bladder and bowel dysfunction as presenting symptoms are relatively uncommon except for tumors that involve the conus medullaris and filum terminale.
Tumors involving the lumbosacral spine present with a cauda equina nerve root compression syndrome. Patients may have radicular pain in the anterior (L4), lateral (L5), or posterior (S1) thigh with corresponding paresthesias followed by muscle wasting of the glutei, hamstrings, or tibialis anterior muscles. Saddle anesthesia, absent ankle reflexes (S1), or plantar (S2) responses may be present. Impotence and loss of anal or bulbar cavernous reflexes also may occur.
 DIAGNOSTIC WORKUP
DIAGNOSTIC WORKUP
History and Physical Findings
Table 37.2 shows the diagnostic workup for primary tumors of the spinal cord. A meticulous and accurate patient history and physical examination are critical aspects of the initial assessment and can often localize suspected spinal tumors. The neurologic examination should concentrate on testing motor and sensory functions and reflexes. The differential diagnosis of a patient with a spinal cord tumor may include syringomyelia, multiple sclerosis, amyotrophic lateral sclerosis, diabetic neuropathy, viral myelitis, or paraneoplastic syndromes.
A cutaneous sensory level may be definable, although the level of cord compression is a few segments higher than the superior level of sensory loss because of pathway crossing characteristics. Loss of pain and heat and cold sensation below a specific dermatomal level indicates compromise of the spinothalamic pathway in the lateral columns. Impaired posture, gait, and coordination and loss of vibration sense indicate compromise of the posterior spinocerebellar pathways or the posterior columns.
At the level of the lesion, flaccid weakness and loss of tendon reflexes may occur. Below the lesion, the same signs are noticed in acute stages, but spastic paralysis and hyperactive tendon reflexes plus an upward Babinski toe sign ensue in subacute and chronic stages. These findings are consistent with lower and upper motor neuron involvement, respectively. The signs and symptoms of neurologic dysfunction may be asymmetric. In some cases, a classic Brown-Séquard syndrome may be present with ipsilateral loss of motor function and fine touch sensation and contralateral loss of pain and temperature sensation below the level of the lesion.
Autonomic reflexes (e.g., sweating) frequently are increased below the level of the lesion and may encompass the whole body if the lesion is cervical.14 Sweating disappears at the level of the compressed cord. Disruption of urinary and bowel function usually occurs later than sensory and motor dysfunction. Early loss of bladder function, saddle anesthesia, and later pain characterize neoplasms of the conus medullaris and filum terminale.
Radiographic Studies
Although plain films are not typically used as the principal imaging modality for the evaluation of suspected spinal canal tumors, abnormalities can be detected from increased intracanal pressure including erosion of vertebral pedicles, enlargement of the anteroposterior diameter of the bony canal, or scalloping of the posterior wall of the vertebral bodies. Calcification may be seen in extramedullary tumors, especially meningiomas, and less frequently in nerve sheath tumors. Overall, plain radiographs of the spine show abnormalities in approximately 50% of patients with primary spinal canal neoplasms.15–18 Changes are more likely to be detected on plain radiographs in children than in adults, such as kyphoscoliosis or scalloping of the vertebral bodies.4,5,19,20
Myelography, once considered the standard examination in evaluation of the spinal cord and canal, is now used principally in patients who are unable to undergo magnetic resonance imaging (MRI) because of the presence of implanted ferromagnetic materials or for whom images at the level of concern are distorted by the presence of surgical hardware. In this situation, computed tomography (CT) scanning combined with myelography will give better spatial resolution. CT myelography may be particularly beneficial as part of radiotherapy treatment planning in the postoperative setting, where the spinal cord may be obscured by artifact on the MRI.21
FIGURE 37.3. Sagittal magnetic resonance imaging scans of a 30-year-old female with a low-grade astrocytoma involving the cervical spine. A: T1-weighted image demonstrates an intramedullary lesion that expands the cord from the level of the craniocervical junction to the level of C7. The enhancing component extends from mid C2 to C5, and there is an associated cystic component at the cranial aspect of the lesion. B: T2-weighted image demonstrates T2 hyperintensity within the lesion suggestive of edema and hypointensity suggestive of blood products.
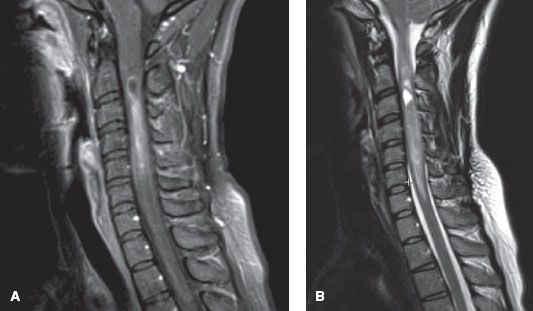
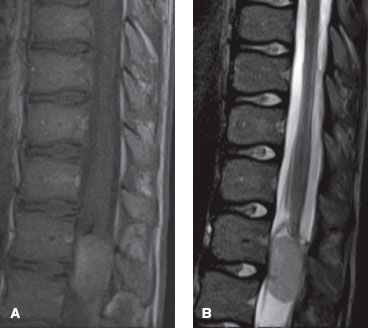
FIGURE 37.4. Sagittal magnetic resonance imaging scans of a 12-year-old male with a myxopapillary ependymoma involving the lumbar spine. A: T1-weighed image demonstrates a homogenously enhancing intradural extramedullary mass at the level of L1. B: T2-weighted image demonstrates hyperintensity of the mass. (Courtesy Aseem Sharma, MD, Mallinckrodt Institute of Radiology.)
Computed Tomography
CT is most helpful in evaluating the spine for extradural pathologic processes. Bone tumors or paraspinal soft tissue masses that secondarily involve the spinal cord (e.g., dumbbell tumors) can be imaged with contrast-enhanced CT scans. Nerve sheath tumors can enlarge the intervertebral foramina or spinal canal and cause smooth erosion of bone. Meningiomas are occasionally calcified. Both of these neoplasms are partially outlined by CSF and produce extramedullary deformity by displacement of the spinal cord.22
Magnetic Resonance Imaging
MRI has replaced myelography and CT as the imaging study of choice in evaluation of tumors of the spinal canal. Sagittal and axial images give a three-dimensional appreciation of the patient’s anatomy and help plan therapy. The various signal characteristics of the CSF—white and gray matter, bone and bone marrow, fat, and flowing blood—all facilitate the interpretation of the study. Some cystic tumors, vascular lesions, or lipomas can be diagnosed based on their characteristic signals on T1- and T2-weighted images without contrast injection. Intravenous gadolinium-diethylenetriamine pentaacetic acid (Gd-DTPA) administration improves the sensitivity of MRI by enhancing the solid component of intramedullary tumors and differentiating them from surrounding edema or syrinx cavities (Figs. 37.3 and 37.4). Unlike low-grade gliomas in the brain, nearly all spinal cord gliomas, regardless of grade, enhance with Gd-DTPA.23 Sagittal T1-weighted images usually localize intramedullary mass neoplasms along with adjacent cysts. Intradural–extramedullary lesions also show enhancement on T1-weighted images after administration of Gd-DTPA. The use of Gd-DTPA also increases the sensitivity of detecting leptomeningeal metastases.23
MRI of the brain should be performed in patients with ependymomas or high-grade astrocytomas to exclude the possibility of neuraxis seeding or the presence of an intracranial primary tumor.
Cerebrospinal Fluid
A patient suspected of having a spinal canal neoplasm should not be subjected to a lumbar puncture before MRI. Symptoms may be exacerbated after a spinal tap because of shifting of the spinal cord and incarceration before the tumor can be localized adequately.20 The CSF usually has increased protein levels and may exhibit xanthochromia, especially with extradural compression conditions, but lower values can be found in cases of intramedullary disease and with compression in the cervical region.14 The incidence of leptomeningeal spread in patients with primary spinal cord glioma is low overall but is substantially more common with malignant tumors. Leptomeningeal dissemination has been reported to be as high as 60% for intramedullary glioblastoma.24,25 Cytological examination of the cerebrospinal fluid should be done in patients with ependymomas (especially anaplastic and myxopapillary types) and high-grade astrocytomas.
Tissue Diagnosis
Suspected primary tumors of the spinal cord and spinal canal must be pathologically confirmed in all circumstances. Strong consideration should also be given to biopsy of any presumed metastatic tumors if they are the first site of disease recurrence after successful management of a previous malignant primary tumor. In rare circumstances in which emergency radiation therapy is indicated to relieve spinal cord compression in the absence of a confirmed cancer diagnosis, a patient should be made aware of the consequences of treatment in the absence of a definitive diagnosis, including the inability to tailor therapy based on histology and possible delay of appropriate treatment for nonmalignant etiologies.
 PATHOLOGIC CLASSIFICATION
PATHOLOGIC CLASSIFICATION
The current comprehensive classification system of spinal tumors was published by the WHO in 2007. It groups the CNS neoplasms by the tumor type and provides a grading system to predict prognosis and guide therapy. Tumor types include neuroepithelial (i.e., astrocytoma, ependymoma, ganglioglioma), nerve sheath (i.e., schwannoma, neurofibroma), meningioma, lymphoma, germ cell, and metastatic tumors.26 Clinically, they are also characterized by their location as either extradural, intradural–extramedullary, or intramedullary.
Intramedullary Tumors
Most intramedullary tumors of the spinal cord are glial in origin, with astrocytomas and ependymomas accounting for the majority.
Astrocytomas are the most common intramedullary spinal cord tumors, comprising approximately 45% of reported cases in adults. In contrast to intracranial astrocytoma, the majority of intramedullary astrocytomas are low grade (WHO grade I or II), including 75% in adults and 85% to 90% in children.27,28,29 Juvenile pilocytic astrocytomas (grade I) in particular, as in other locations in the CNS, are not infiltrative in nature. Recognition of this feature, along with advances in surgical techniques and intraoperative monitoring, has led neurosurgeons to manage these tumors with more radical resections.19,30,31–32 However, many fibrillary astrocytomas (grade II) as well as anaplastic astrocytomas (grade III) and glioblastoma multiforme (grade IV) are infiltrative, and complete resection carries a significant risk of neurologic disability. In these cases, a subtotal resection or biopsy may be the only safe surgical option.
Astrocytomas are more likely to occur in the cervical and thoracic regions than ependymomas. Low-grade astrocytomas are typically confined to a focal segment of the spinal cord. Rarely, astrocytomas may involve a large part of the spinal cord, at which point they are designated a holocord tumor. These holocord tumors may result entirely from tumor involvement or from a large syrinx in addition to the original tumor. The term “holocord” must be used with caution, however, as many of the previously reported cases were made by myelography, and recent series using MRI suggest a significantly decreased frequency of these tumors.33
A discriminating feature of intramedullary spinal cord tumors is the presence or absence of a syrinx. Syringes associated with spinal cord astrocytoma occur in 20% to 40% of patients.34 Syringes can span the entire length of the cord, although they tend to favor more rostrally located tumors, as well as favoring the more rostral portion of the tumor itself.34 In certain instances, a syrinx may even extend into the medulla, producing an obstructive hydrocephalus.35 The syrinx itself can be responsible for significant neurologic deficit.
Approximately 40% of intramedullary tumors are ependymomas. Ependymomas are derived from glial cells similar to those lining the ventricular system. Several histologic types have been reported and include cellular, epithelial, tanycytic, subependymoma, myxopapillary, or mixed types. Cellular (classic) ependymomas (WHO grade II or III) typically develop in the cervical and thoracic spinal cord. Myxopapillary ependymomas (grade I) often arise from within the filum terminale and typically occur in the lumbosacral region.36–38,39–40 Myxopapillary ependymomas frequently can be completely excised. In many circumstances, however, these tumors tightly envelope the nerve roots of the cauda equina, making en bloc excision difficult. Gross total resection of these tumors often requires piecemeal removal. The myxopapillary variant may be biologically less aggressive than the cellular variant, but late recurrences can occur even after complete gross excision; therefore, long-term follow-up of these patients is required.40–44
A variety of vascular neoplasms can arise from within the spinal cord, including arteriovenous malformations, hemangiomas, and hemangioblastomas. Approximately one-third of hemangioblastomas are associated with von Hippel-Lindau disease. These are benign neoplasms that are usually well circumscribed and amenable to surgery.45 Unresectable or recurrent hemangioblastomas may be treated with stereotactic radiosurgery.46
Intradural–Extramedullary Tumors
Most intradural–extramedullary neoplasms are meningiomas, nerve sheath tumors, or myxopapillary ependymomas. They usually are amenable to complete surgical excision.
Meningiomas are usually benign, well-encapsulated neoplasms that are easily separated from the spinal cord; most can be completely excised, and they rarely recur. They may arise anywhere within the intradural space but are found in the thoracic region in approximately 80% of patients.47–49 Meningiomas are uncommon in the lumbar region and rare in the sacrum. At least 80% of meningiomas occur in women 40 years of age or older.47,48
Nerve sheath tumors arise from the Schwann cell, the cell responsible for insulating peripheral nerves and contributing to impulse conduction. Nerve sheath tumors have been called, neurofibroma, schwannoma, neuroma, and neurilemoma in the past. More recently, a distinction between neurofibroma and schwannoma has been made. Although both tumors arise from Schwann cells, certain gross, microscopic, and clinical features help distinguish the two.29 Neurofibromas typically encase involved nerve roots, while schwannomas commonly displace the nerve roots due to their asymmetric growth. The plexiform neurofibroma is associated with type 1 neurofibromatosis, and the presence of multiple tumors helps establish the diagnosis of this genetic condition. In contrast, schwannoma is associated with type 2 neurofibromatosis. Patients with type 1 neurofibromatosis may be at risk for malignant tumor transformation following radiotherapy.50 Nerve sheath tumors usually are solitary and may occur in any section of the spinal canal. They are evenly distributed in the cervical, thoracic, and lumbar regions; they are least common in the sacrum. They occur in men and women with equal frequency and are most commonly diagnosed in the fourth through sixth decades of life. Most of these tumors are completely intradural, although 10% to 15% may have an extradural component as well (so-called dumbbell tumors). Most nerve sheath tumors are benign, well-encapsulated lesions that are amenable to total surgical excision. The rare malignant nerve sheath tumors have a natural history similar to soft tissue sarcomas, and they should be treated as such.51–53
Miscellaneous Neoplasms
Unusual intradural–extramedullary tumors include lipomas, dermoids, and epidermoid tumors. They are typically benign and amenable to complete resection. Even if incompletely excised, recurrences are usually slow.
Extradural Tumors
Most extradural tumors are metastatic, and the presentation and management of these is discussed in Chapter 94. A variety of primary bone and soft tissue tumors may arise from an extradural location and involve the spinal canal. Bone tumors include osteosarcoma, chordomas, chondrosarcoma, and Ewing’s sarcoma. Soft tissue tumors include soft tissue sarcomas, including malignant nerve sheath tumors, lymphomas, and neuroblastomas. These tumors and their management are discussed in Chapters 78 (lymphoma), 82 (osteosarcoma/chordoma), 83 (soft tissue sarcoma), 86 (neuroblastoma), and 88 (Ewing’s tumor), respectively.
 PROGNOSTIC FACTORS
PROGNOSTIC FACTORS
The major prognostic factors in patients with primary spinal canal tumors are tumor type and grade, tumor extent and location, patient age, and presenting neurologic function. Treatment-related factors that influence the outcome include tumor resectability and the use of radiation therapy for certain tumor types. Many of these factors are interdependent. For example, ependymomas occur most frequently in the distal spinal canal and are more often resectable than astrocytic tumors.
Recently, Milano et al.54 analyzed 664 patients with spinal cord astrocytomas and 1,057 patients with spinal cord ependymomas using the SEER database. In this largest study to date on prognostic factors for long-term outcome, lower grade, younger age, and surgical resection were associated with significantly better overall survival and cause-specific survival for both astrocytomas and ependymomas. Radiotherapy was associated with worse cause-specific survival, but this is likely due to adverse selection bias. The 5-year overall survival of grades 1, 2, 3, and 4 astrocytomas was 82%, 70%, 28%, 14% and the 5-year cause-specific survival was 89%, 77%, 36%, 20%, respectively. The 5-year overall survival of grades 1, 2, and 3 ependymomas was 92%, 97%, and 58% and the 5-year cause-specific survival was 100%, 98%, and 64%, respectively.
Several investigators have reported that patients with rostral tumors have a worse survival and neurologic outcome than patients with more caudal tumors.8,55–57 Guidetti et al.56 stated that patients with cervical lesions had a higher surgical risk and complication rate, which made thorough resection of tumors in this location difficult and sometimes inadvisable. In a series of 62 patients with exclusively intramedullary ependymomas, patients with high cervical presentations (above C5) accounted for 4 of 6 postoperative deaths because of apneic respiratory complications.57 In the Mallinckrodt Institute of Radiology experience, Garcia8 reported that the primary tumor location was the most important prognostic feature. It was suggested that a greater concentration of function per unit volume of the upper spinal cord compared with that of the cauda equina accounted for the worst neurologic outcome and survival in patients with rostral tumors. Chun et al.,55 from the Medical College of Virginia, also reported that patients with cervical lesions had significantly worse outcomes than patients with tumors in other sites. In both the Mallinckrodt Institute of Radiology and the Medical College of Virginia experiences, tumors affecting the rostral or cervical spinal cord were more likely to be astrocytomas, and tumors in the caudal spinal cord, filum terminale, or cauda equina were more likely to be ependymomas. The anatomic dependence of various tumor types may also contribute to the better prognosis seen in patients with tumors of the lower spinal canal.
Extensive involvement of the spinal cord with an ependymoma is associated with a worse outcome. Linstadt et al.10 reported a 93% 10-year disease-specific survival rate with localized ependymoma compared with 50% for patients with diffuse tumors. Extensive tumors have a 50% local failure rate after surgery and radiation therapy, compared with only 20% for limited disease (one to three vertebral body segments).39 However, extent of disease has not been a prognostic factor in other series.58 Myxopapillary ependymomas that most commonly involve the cauda equina are felt to be less aggressive than other ependymomas,40–42 but they have been reported to seed the CSF.59,60 Encapsulated myxopapillary tumors of the cauda equina are frequently amenable to complete en bloc excision, and the recurrence rate is very low. Unencapsulated or adherent tumors often are removed piecemeal and are associated with a high local recurrence rate after surgery alone.39,40,43,61,62
Neurologic function at diagnosis is an important clinical prognostic factor. In general, the fewer the symptoms and the better the neurologic function at presentation, the greater the likelihood the tumor will be controlled with fewer long-term adverse neurologic sequelae.8,31,56,57,63 Poor neurologic function in patients with spinal cord tumors is often attributable to the disease process and a prolonged delay in diagnosis rather than the effect of surgery or radiation therapy.4,27,28,61,64,65
 SURGICAL MANAGEMENT
SURGICAL MANAGEMENT
Intramedullary Tumors
Intramedullary tumors, most of which are astrocytomas and ependymomas, present a surgical challenge. Complete surgical excision is the treatment of choice if it can be achieved without compromising neurologic function. Ependymomas are more frequently amenable to gross total excision than are astrocytomas.30,56,66 Complete resection of intramedullary tumors with preservation of neurologic function was not possible until 1940, when Greenwood20 introduced the bipolar coagulation forceps. Since then, other technological advancements have emerged, including the dissecting microscope, intraoperative ultrasound, ultrasonic aspirator, contact laser scalpel, and intraoperative neurophysiologic monitoring.67 These modern surgical techniques have increased the complete resectability of intramedullary tumors while minimizing neurologic injury.68–70
Intraoperative ultrasonography is used to localize the lesion, define its extent, and assess the progress of tumor resection.71 The ultrasonic aspirator allows removal of tissue fragments from within 1 mm of the vibrating tip, permitting dissection immediately adjacent to vital neural tissue.72 The contact laser scalpel only delivers thermal energy to tissue upon direct contact, and the laser beam resides entirely within a coated sapphire crystal probe tip. It provides precise dissection of tumor with little mechanical or thermal injury to normal spinal cord parenchyma.73
Intraoperative neurophysiologic monitoring is considered standard of care for resection of intramedullary tumors. It is dependent on two key components: somatosensory-evoked potentials (SEPs) and motor-evoked potentials (MEPs). SEPs are monitored continuously during the incision of the dorsal midline of the spinal cord to avoid injuring the dorsal column, as the cord anatomy is often distorted by the tumor. MEPs are monitored once the surgeon starts to dissect the tumor to avoid irreversible damage to the corticospinal tract. MEPs should be monitored using a combination of epidural electrodes (D waves) and signals from limb muscle (mMEPs). Typically, a decrement of 50% or more of D-wave amplitude is considered a major indication to stop surgery.74 Sala et al.75 reported that the use of intraoperative neurophysiologic monitoring during resection of intramedullary tumors significantly improved long-term ambulatory status as compared to historical controls.
The risk of paralysis after surgery is less than 1% of patients with minimal or no preoperative neurologic deficits, but may be much higher for those who present with more substantial deficits. Approximately one-third of patients will develop temporary motor deficits after surgery and most will have at least some postoperative deterioration in neurologic status.76,77 If complete excision of low-grade spinal cord tumors is achieved, the local recurrence rate is low and prognosis is excellent without additional adjuvant therapy. In patients who recur, tumor regrowth is often slow and second resection may be possible.78–80
Intradural–Extramedullary Tumors
The treatment of choice for most tumors in this location is maximal surgical excision with preservation of neurologic function. Most benign nerve sheath tumors and meningiomas can be completely resected using a posterior approach with a standard posterior laminectomy.29 Nerve sheath tumors rarely recur after satisfactory surgical removal. In contrast, as much as 15% of spinal meningiomas recur as late as 10 years after gross total or near total removal.81 Piecemeal resection of ependymomas of the filum terminale can be accomplished with little neurologic disability; however, the risk of recurrence in these patients is significant, and adjuvant radiation therapy is warranted.39,40,62,82,83
In young children, posterior laminectomy is being abandoned and replaced with posterior osteoplastic laminotomy. Replacing the posterior bony elements of the spinal canal is less likely to cause significant kyphotic deformity and affords better protection of the spinal cord.69,84,85
 CHEMOTHERAPY
CHEMOTHERAPY
The reported use of chemotherapy for primary spinal canal tumors is limited. Outside of a clinical trial, chemotherapy is often reserved for patients with progression of disease following surgery and radiotherapy with no other treatment options. Combined with the low incidence of the disease, most studies are retrospective with very few patients, which limits any definitive conclusion regarding their efficacy. The use of chemotherapy for spinal canal tumors is often extrapolated based on the experience for intracranial tumors. However, such assumption requires further validation as the underlying oncogenesis and biological pathways may be different between the two entities.86
Platinum and etoposide are generally considered the most active agents for ependymomas. In the setting of recurrent ependymoma, platinum-based chemotherapy has been shown to produce higher response rates than nitrosourea-based chemotherapy, but most patients in the studies had intracranial ependymomas.87,88 In a prospective phase II study, 10 consecutive patients with recurrent cellular spinal cord ependymomas were treated with oral etoposide. Two patients had partial responses, and five patients had stable disease. The median overall survival was 17.5 months.89
Temozolomide is an attractive agent for primary spinal cord glioma given its success in treating intracranial astrocytoma.90 Kim et al.91 reported their experience of treating two patients with primary spinal cord glioblastoma multiforme with concurrent radiotherapy and temozolomide followed by adjuvant temozolomide. The two patients survived 12 and 16 months, respectively, in contrast with a median survival time of 9 months in other reports. Chamberlain92 reported a series of 22 patients with recurrent WHO grade II glioma treated with temozolomide. Overall, there were 18% partial response and 55% with stable disease. The median survival was 23 months and progression-free survival at 2 years was 27%.
For young children, especially those less than 3 years of age, there is an even greater interest in identifying effective and minimally toxic chemotherapy to delay or eliminate radiation therapy. There are two prospective cooperative group trials that have included children with primary spinal cord astrocytomas. In one clinical trial conducted by the French Society of Pediatric Oncology, eight children with unresectable or recurrent intramedullary low-grade gliomas were treated with a planned 16-month course of carboplatin, procarbazine, vincristine, cyclophosphamide, etoposide, and cisplatin. Seven of the patients had a clinical or radiographic response to the chemotherapy. Five of the patients remained progression free, with follow-up ranging from 16 to 59 months.93 In the Children’s Cancer Group 945 trial, 13 children with high-grade astrocytic spinal cord neoplasms were assigned to receive two cycles of “8-drugs-in-1-day” chemotherapy before radiation therapy, then eight additional cycles thereafter. At 5 years, 46% of the children had no progression and 54% were alive. The authors argued that more intensive therapy was necessary.94 Mora et al.95 recently reported a small but thought-provoking study of three infants with spinal cord astrocytomas (WHO grade II or III) treated with irinotecan and cisplatin. All three infants had subtotal resection and progressed on conventional carboplatin-based chemotherapy. After switching to irinotecan and cisplatin, all three patients had remarkable radiologic and clinical response. At the time of the last follow-up, they had remained in remission at 12, 20, and 48 months after diagnosis. The authors hypothesized that irinotecan and cisplatin may provide synergistic effect without overlapping toxicities. Currently, children with high-grade astrocytomas of the spinal cord are eligible to enroll in a phase II Children’s Oncology Group clinical trial of adjuvant radiation and concurrent temozolomide followed by additional temozolomide and lomustine chemotherapy for high-grade gliomas (COG-ACNS0423).
 RADIATION THERAPY
RADIATION THERAPY
There are currently no randomized controlled trials to guide the role of radiation therapy for primary spinal canal tumors. Clinical use of postoperative radiation therapy is generally guided by patterns of failure and the prevailing attitude of many radiation centers.96 Patients with completely resected low-grade astrocytomas19,29,31,32,97 and ependymomas78,80,98–100 typically have an excellent prognosis, with local failure rates of less than 10% without additional therapy. Most centers do not advocate routine use of adjuvant radiation therapy in this setting, but long-term follow-up is indicated as late failures can occur.
In contrast, adjuvant radiation therapy after incomplete resection or piecemeal excision of low-grade ependymomas and astrocytomas is supported by retrospective analyses. Guidetti et al.56 first reported a beneficial outcome in patients receiving radiation therapy after an incomplete excision of an ependymoma. Patients who have undergone complete excision of a cauda equina ependymoma by piecemeal removal have a local failure rate that ranges from 20% to 43%.39,40,62 The addition of radiation therapy in patients who have undergone piecemeal excision of a cauda equina ependymoma produces a local recurrence rate equal to that of patients undergoing gross total resection.39,62,83 In a multi-institutional series, adjuvant radiation therapy significantly improved progression-free survival in the 40 patients with low- and intermediate-grade astrocytomas.30 After adjuvant radiotherapy, the cumulative incidence of local failure ranges from approximately 20% for low-grade ependymoma to 40% for low-grade astrocytoma.96
Nonetheless, there are clinical circumstances in which careful follow-up after incomplete resection is appropriate, with a second surgery or radiation therapy considered at the time of progression or recurrence. Radiation therapy of the spine in a child may produce a spinal deformity (i.e., scoliosis or kyphosis) because of retardation of bone growth from damage to epiphyseal plates of the vertebral bodies as well as soft tissue fibrosis and contracture.101 Most spinal cord tumors in young children are either low-grade astrocytomas or well-differentiated ependymomas that have a very low growth rate. Delaying radiation therapy until recurrence or tumor progression may allow the child to grow at a normal rate for several years before receiving radiation therapy. Constantini et al.102 reported a series of 164 patients younger than 21 years of age treated with radical surgery alone without adjuvant radiotherapy. Gross total resection or subtotal resection was achieved in 77% and 20% of patients, respectively. The 3-month neurologic function was 60% stable, 16% improved, and 24% deteriorated compared with preoperative function. The 5-year progression-free survival rate for low-grade and high-grade tumors was 78% and 30%, respectively.
The prognosis of high-grade astrocytomas and ependymomas is dismal, and multimodality treatment is recommended, ideally on a clinical trial. Adjuvant radiation therapy is routinely recommended regardless of the extent of resection. The role of concurrent and adjuvant chemotherapy for high-grade intramedullary astrocytoma remains inconclusive as discussed earlier. Despite aggressive treatments, few patients with high-grade astrocytomas survive beyond 2 years.96,103 As with intracranial high-grade glioma, novel therapies beyond traditional radiotherapy and cytotoxic chemotherapy are desperately needed.
Relapse can occur years after treatment and is predominantly local. In a series of 37 spinal ependymomas treated with adjuvant radiotherapy, more than 50% of failures developed 5 years after diagnosis, and the extent of surgical resection correlated with time to progression.104 These findings were confirmed in another series of patients with spinal myxopapillary ependymoma, where Chao et al.105 reported a median time to recurrence of 7.7 years.
The prognosis is excellent for most patients with intradural–extramedullary tumors, which rarely recur after total excision. However, subtotally resected meningiomas may recur late after surgery.81 Some investigators have advocated postoperative radiation therapy using either conventional fractionated external-beam radiotherapy or stereotactic radiosurgery.106,107 Radiation therapy is beneficial to patients undergoing subtotal resection or piecemeal excision of intradural–extramedullary ependymomas.10,39,40,62 Data supporting the routine use of radiation therapy in the management of patients with nerve sheath tumors, vascular malformations, lipomas, hemangiomas, teratomas, and dermoids are limited.
 RADIATION THERAPY TECHNIQUES
RADIATION THERAPY TECHNIQUES
Target Volume
Historically, it had been recommended that superior and inferior field borders encompass two vertebral bodies above and below a tumor defined by myelography, with the width of the field approximated between the tips of the lateral processes of the vertebral bodies. With the advancement of CT simulation and MRI fusion, the gross tumor volume (GTV) can be more accurately defined and smaller margins may be used. For low-grade astrocytomas or ependymomas, a clinical target volume (CTV) margin of 0.5 to 1 cm is appropriate. The CTV should encompass the preoperative GTV plus any associated intratumoral cysts. It is not necessary to include an intramedullary syrinx that extends above or below the primary tumor unless there is radiographic or surgical evidence of tumor extension to these regions. Planning treatment volume (PTV) margins of 0.5 cm or less may be used to account for setup error, with smaller PTV margins necessitating adequate patient immobilization and optimal daily localization techniques (orthogonal kilovoltage imaging, cone-beam CT, etc.).
High-grade astrocytomas and ependymomas can be more infiltrative, and a larger CTV margin of at least 1.5 cm craniocaudally should be used. Merchant et al.108 described a diffuse failure pattern in children with high-grade gliomas shortly after completing radiation therapy, suggesting that the tumor was not adequately covered in the irradiated volume and the need for larger CTV margins of 1.5 cm. The intervertebral foramina should be included within the CTV if tumor extension is suspected.
For myxopapillary ependymomas involving the conus, a 1.5-cm CTV margin cephalad and caudad to the GTV is used, but not beyond the thecal sac, which is typically at the level of S2-3. If the cauda equina is involved, the CTV should extend inferiorly to encompass the entire thecal sac, with the volume widened at the sacroiliac joints to ensure adequate coverage of the meningeal sleeves in the intervertebral foramina. Failure to adequately encompass the thecal sac has been associated with an increased rate of treatment failure.62
Craniospinal or spinal axis irradiation usually is not indicated in the treatment of most spinal cord tumors; local failure accounts for most tumor recurrences.8,10,30,39,97,109 However, neuraxis dissemination may be seen in patients with anaplastic ependymomas,110 malignant astrocytomas,24,111 and myxopapillary ependymomas.59,60 Craniospinal irradiation may be considered in these situations.
Radiation Technique
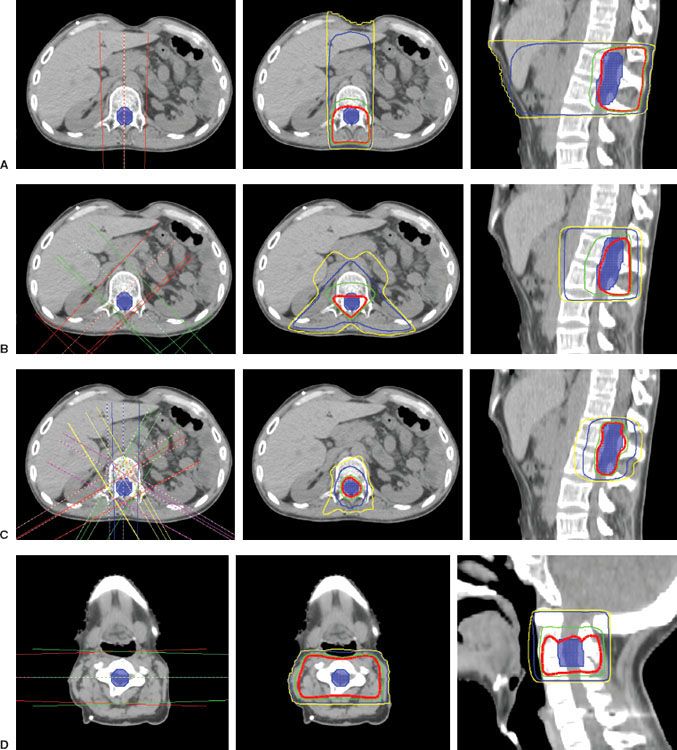
For conventional external-beam field arrangements, cervical cord tumors are typically treated with parallel opposed lateral fields to reduce dose to the oral cavity, larynx, and pharynx. Thoracic cord tumors are commonly treated with direct posterior or posterior wedge fields to limit dose to the anterior structures such as the lungs, esophagus, and heart. Lumbar and cauda equina tumors are mostly treated with opposed anteroposterior–posteroanterior portals because of the lumbar lordosis and the deep location of the vertebral canal. Conformal or intensity-modulated radiation therapy (IMRT) methods may reduce normal tissue toxicity60 and should be considered when exit dose to the anterior midline structures of the trunk would otherwise be excessive (Fig. 37.5). However, IMRT may increase the volume of normal tissue receiving low doses and is theoretically associated with a higher risk of secondary malignancy.112
In female patients requiring treatment to the lumbosacral spine for cauda equina tumors, a lateral technique may be used to avoid exit irradiation to the ovaries and uterus. This technique prevents the anterior pelvic structures from receiving significant irradiation dose, which is desirable in young women and girls to minimize incidental irradiation of the ovaries. The superior aspect of this field can be matched to the divergence of a superior posteroanterior field in a fashion similar to the junction of a cranial portal to a spinal portal in craniospinal irradiation. Beam modifiers such as wedges or tissue compensators may be required with this lateral beam arrangement. Care should be taken to avoid irradiating the kidneys at the L1 through L3 levels with this technique. Arms should be positioned appropriately to avoid entrance or exit irradiation from the lateral beams.
The depth of the vertebral surface of the cord beneath the skin surface is determined from CT or MRI, and for short field lengths this depth is used for dose prescription. The treatment plan should provide a homogeneous dose distribution. For small lesions of the cervical spinal cord, where lateral fields will be used, radiation beam energies of 4 to 6 MV photons achieve a homogeneous dose distribution. Lesions involving the thoracic and lumbar spine often require combinations of low-energy (4 to 6 MV) and high-energy (18 to 25 MV) photons to achieve a homogeneous dose distribution when posterior fields are used. Attention to the exit dose delivered to anterior anatomical structures needs to be considered against dose heterogeneity in the target volume. Parallel-opposed posterior and anterior fields or paired oblique wedge fields can give homogeneous dose distributions with x-ray energies as low as 4 or 6 MV.
Radiation Dose
Low-grade astrocytomas and ependymomas should be irradiated to a total dose of 50.4 Gy, given in 1.8 Gy daily fractions. High-grade astrocytomas can be treated to a dose of 54 Gy with 1.8 Gy daily fractions. High-grade ependymomas and multifocal low-grade astrocytoma should be treated to a dose of 50.4 to 54 Gy.96 Limited dose response data exist for spinal cord tumors. In the Mallinckrodt series of 37 patients with primary spinal cord tumors, local control and survival were significantly better for those received 40 Gy or higher.8 Shaw et al.39 reported that the local failure rate of ependymomas was 35% in patients receiving 50 Gy or less compared with only 20% in patients receiving more than 50 Gy. However, doses beyond 50.4 Gy have not been shown to improve local control or survival.10,66,113 In patients with high-grade ependymomas or other spinal cord gliomas with evidence of CSF dissemination, craniospinal irradiation should be considered. Typical doses to the craniospinal axis range from 36 to 45 Gy, with a boost to sites of gross tumor to 50.4 to 54 Gy.
FIGURE 37.5. Treatment planning for spinal cord tumors. Planning treatment volume (PTV) in solid blue. Red line, prescription dose of 50.4 Gy; Green line, 95% of prescription dose of 47.88 Gy; Blue line, 30 Gy; Yellow line, 20 Gy. A: A single posteroanterior field. The advantages of this beam arrangement include simplicity and near-universal applicability in most spinal cord radiation therapy treatments. One disadvantage of this field arrangement is the large volume of tissue that receives a significant exit dose. The axial and sagittal isodose displays reflect a 6-MV x-ray beam treated to a point just anterior to the PTV. B: Paired posterior oblique wedge fields. The advantage of this technique is a decrease in high exit-dose irradiation to anterior tissues with a more conformal irradiation dose distribution near the target volume. Disadvantages include more complicated treatment setup and verification. The axial and sagittal isodose displays reflect a 45-degree wedged pair of 6-MV x-ray beams treated to the same point with a 90-degree hinge angle. C: Intensity-modulation radiation therapy (IMRT). An advantage is a highly conformal dose distribution with excellent sparing of adjacent critical structures. Disadvantage are complexity and high integral dose. The axial and sagittal isodose displays reflect a five-field static IMRT plan with 6-MV x-rays. D: Opposed lateral fields. An advantage is a homogeneous dose distribution in the target volume with sparing of anterior structures from significant irradiation dose. A disadvantage is limited applicability in cervical and lower lumbosacral sites. Exclusive use of this field arrangement in the thorax and upper abdomen is inappropriate because of limited lung and kidney tolerance. The axial isodose display reflects a pair of laterally directed 6-MV x-ray fields treated to the midplane of the cervical spine. (C, Courtesy Andrew Lindsey, Washington University in St. Louis Department of Radiation Oncology.)