t(1:13)(p36;q14)
PAX7-FOXO1A
CREB1-EWSR1
COL1A1
IGF2
t(21;22)(q22;q12)
t(7;22)(p22;q12)
t(17;22)(q12;q12)
t(2;22)(q33;q12)
ERG-EWSR1
ETV1-EWSR1
E1AF-EWSR1
FEV-EWSR1
t(9;17)(q22;q11)
t(9;15)(q22;q21)
NR4A3-RBP56
NR4A3-TCF12
PDGFRA
t(11;16)(p11;p11)
CREB3L1-FUS
t(2;19)(p23;p13)
t(2;17)(p23;q23)
t(2;2)(p23;q13)
ALK-TPM4
ALK-CLTC
ALK-RANBP2
t(12;22)(q13;q12
DDIT3-EWSR1
SSX2-SYT
SSX4-SYT
amplification
CDK4
SAS
HMGA2
LOH, Loss of heterozygosity.
Genetics and Molecular Pathogenesis
SS contain a specific translocation between chromosomes X and 18 [t(X;18);(p11.2;q11.2)]. 4 This translocation results in fusion of the SYT gene on chromosome 18 with either SSX1 or SSX2 (or rarely SSX4) on chromosome X. The resulting fusion gene contains SYT gene minus the final 8 C-terminal amino acids and the C terminus of the SSX gene (see Figure 44-1, C).
In the literature to date, it appears that biphasic SS much more commonly carries the SYT-SSX1 fusion, whereas monophasic SS shows either SYT-SSX1 or SYT-SSX2. 11–13 This trend between fusion gene product and histologic type suggests the possibility that the SYT-SSX1 fusion gene is more efficient at promoting epithelial differentiation than the SYT-SSX2 fusion gene. However, some biphasic SS carry the SYT-SSX2 rather than the SYT-SSX1 transcript, so this reported association is by no means consistent.
Attempts have also been made to correlate fusion type with prognosis, although the results are conflicting. Several studies suggested that patients with the SYT-SSX2 translocation have a better prognosis than those with the SYT-SSX1 transcript; 13,14 others have found no statistically significant correlation between fusion type and prognosis. 12 All of these studies were retrospective, and none could control for differences in treatment or selection bias in follow-up data. 15 The proposed correlation of fusion gene type with prognosis therefore remains unconfirmed.
SYT is widely expressed during early murine embryogenesis and in the adult. 11 The gene encodes a protein with three possible SH2- and one possible SH3-binding domains (likely protein-protein interaction domains), a novel N-terminal domain (termed an SNH domain), and a C-terminal domain rich in glutamine, proline, glycine, and tyrosine (QPGY domain) similar to those found in the EWSR1 gene and other transcriptional activators (see Figure 44-1, C). There is in vitro evidence that SYT can act as a transcriptional activator and SYT has been shown to bind to the SWI/SNF chromatin remodeling complex. 16 SYT also interacts with the p300 nuclear pore protein, and this interaction appears to promote cell-cell adhesion. 17 An SYT deletion mutant lacking the eight C-terminal amino acids (the most common SYT mutant found in the SYT/SSX fusion gene product) acts in a dominant negative fashion, preventing cell adhesion to an extracellular matrix. 17
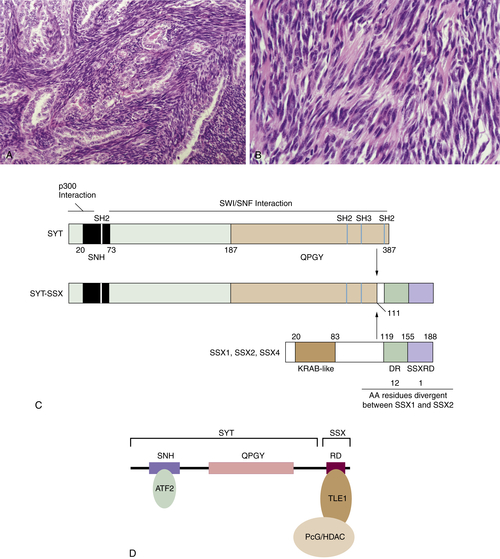
Figure 44-1 Synovial sarcoma (A) Biphasic synovial sarcoma, with glandular differentiation within a spindle cell background. (B) Monophasic synovial sarcoma, demonstrating the typical densely packed spindle cells growing in a fascicular pattern within a background of wiry stromal collagen. (C) Diagram of SYT, SSX, and the SYT-SSX fusion proteins. (D) Interaction of SYT-SSX with ATF2 and TLE1 leads recruitment of PcG/HDAC via TLE1. AA, Amino acids; DR, SSX divergent region; KRAB, Kruppel-associated box; QPGY, SYT glutamine, proline, glycine, and tyrosine-rich domain; SNH, SYT N-terminal domain; SSXRD, SSX repressor domain.
SSX1 and SSX2 share significant homology (81% identity) to each other and belong to a gene family whose expression is predominantly restricted to germ cells and tumors. 11 The SSX protein contains an N-terminal domain similar to the Kruppel-associated box (KRAB) domain found in several transcriptional repressors, and an acidic C-terminal region (SSX-RD) that appears to be a novel transcriptional repressor domain 18 required for colocalization with polycomb group (PcG) proteins, which are important for maintaining transcriptional repression from one cell cycle to the next. 19
The transforming capability of the SYT-SSX1 fusion gene product has been demonstrated in vitro and in vivo. 20 The N-terminal region of SYT (which binds to the SWI/SNF chromatin remodeling complex) is required for the transforming properties of SYT-SSX1, suggesting a role for transcriptional regulation by SYT-SSX1 via the SWI/SNF complex in SS tumorigenesis. Recently, proteins involved in the transcriptional regulatory activity of SYT-SSX have been identified, including activating transcription factor 2 (ATF2; a transcriptional activator), TLE1 (a transcriptional repressor), and histone deacetylases (HDAC). 21 TLE1 is strongly expressed in SS and is a sensitive diagnostic marker for SS. 22–24 Knockdown of both ATF2 and TLE1 in both human and mouse SS cell lines via small interfering RNA (siRNA) prevented tumor cell colony formation and inhibited cell growth, 21 supporting a role for these proteins in the oncogenic activity of SYT-SSX in SS. The association of SYT-SSX with ATF2 and TLE1 results in repression of ATF2 target genes via recruitment of HDAC/PcG by TLE1 (see Figure 44-1, D).
The role of the HDAC/PcG complex in the oncogenic effects of SYT-SSX provides a clinically relevant therapeutic target, namely, HDAC inhibitor targeted therapy. In preclinical studies, HDAC inhibitors have shown early success in SS growth inhibition and remain an area of significant interest in SS treatment. 21,25,26
In addition to a potential role for HDAC inhibitors in SS targeted therapy, recent data have implicated a role for the phosphatidylinositol-3′-kinase (PI3K)/AKT signaling pathway in SS tumorigenesis, 27 as this signaling is frequently activated in SS. Inhibition of PI3K also decreased SS growth in vitro. 27 These data point to a potential role for PI3K inhibitors in SS therapeutic strategies.
Atypical Lipomatous Tumor/Well-Differentiated Liposarcoma
Clinical Description and Pathology
Liposarcoma as a class is the most common malignant soft tissue neoplasm. Of the several distinct subtypes, atypical lipomatous tumor/well-differentiated liposarcoma (ALT/WDL), a sarcoma of intermediate (locally aggressive) malignancy, is the most common. 28 It is a tumor of older adults, most often presenting in the sixth or seventh decades. Males and females are equally affected. ALT/WDL occurs most frequently in the extremities or retroperitoneum, followed by the paratesticular region, mediastinum, and head and neck. They tend to be deep-seated, slowly growing masses, and thus are often quite large before coming to clinical attention.
Anatomic location is the most important prognostic factor, because ALT/WDL does not metastasize unless dedifferentiation has occurred (see later discussion). Indeed, although the terms ALT and WDL are synonymous, tumors in surgically resectable locations in the limbs and trunk are labeled ALT because wide excision is curative. In contrast, those located in the retroperitoneum and mediastinum, where wide excision is difficult, are referred to as WDL, because repeated and uncontrolled local recurrences are very common, and mortality is high even in the absence of dedifferentiation.
Histologically, ALT/WDL is divided into four subtypes (without prognostic implications): adipocytic (lipoma-like), sclerosing, inflammatory, and spindle cell, of which the first two are the most common. 28 In general, ALT/WDL is composed of relatively mature adipose tissue with significant variation in cell size and varying degrees of nuclear atypia in adipocytes and stromal cells, particularly within fibrous septa. Lipoblasts are often present; however, they are frequently rare and are not required for the diagnosis. Sclerosing ALT/WDL is characterized by collagenous stroma containing pleomorphic hyperchromatic stromal cells and is most common in the retroperitoneum and paratesticular region. Inflammatory ALT/WDL is rare but is important to recognize in that it can be misdiagnosed as Hodgkin’s lymphoma, as other sarcomas, or even as a nonneoplastic process, because of the very extensive chronic inflammation present. Spindle-cell ALT/WDL is composed of only mildly atypical spindle cells admixed with adipocytes and lipoblasts (in many cases), in a fibrous or myxoid stroma.
Dedifferentiation, defined by progression to nonlipogenic, often morphologically high-grade sarcoma, occurs in approximately 10% of ALT/WDL. It most commonly occurs in the retroperitoneum, because dedifferentiation appears to be a time-dependent (or size-dependent) phenomenon, and retroperitoneal ALT/WDL often remains asymptomatic until it reaches a large size and tends to have a protracted clinical course because of its relative unresectability. Histologically, dedifferentiated liposarcoma (DDLPS) is characterized in most cases by an abrupt transition from ALT/WDL areas to variably pleomorphic spindle cell sarcomatous areas. DDLPS has a 40% to 50% local recurrence rate, a 15% metastatic rate, and a 30% 5-year mortality rate. 29
Genetics and Molecular Pathogenesis
Giant marker and supernumerary ring chromosomes are the hallmark of ALT/WDL and are also present in DDLPS. 30 These giant ring and marker chromosomes contain massive amplification of the 12q13-15 chromosomal region. 31 A number of other chromosomal regions, including 12q21-22 and 1q21-25, have also been shown to be co-amplified. The p53 regulator MDM2, located in 12q14-15, is consistently amplified in ALT/WDL, typically in association with neighboring genes, including CDK4, SAS, and HMGA2. Expression of both MDM2 and CDK4 has been shown to be a sensitive and specific marker to distinguish ALT/WDL from benign lipoma (which can be a challenge, especially if there is coexistent fat necrosis). 32 MDM2 negatively regulates the tumor suppressor p53 by targeting it for ubiquitin-mediated destruction. 33 Approximately 30% to 40% of sarcomas in general display MDM2 overamplification, and 38% of tumors in mice overexpressing MDM2 are sarcomas. 34 Nutlins, a class of small-molecule inhibitors of MDM2, 35 have been shown to have growth inhibitory effects on ALT/WDL cell lines, raising their consideration as a new potential therapeutic option. 36
Although not fully elucidated, recent data have begun to unravel the molecular pathogenesis of tumor progression from ALT/WDL to DDLPS. DDLPS differ cytogenetically from ALT/WDL in that they often show additional complex karyotypic changes, including involvement of 1p32 and 6q23. 37 DDLPS also have been shown to possess a greater degree of 12q amplification than ALT/WDL. Chibon and colleagues 38 identified a role for amplification of ASK1 (MAP3K5) a mitogen-activated protein (MAP) kinase kinase on 6q23 that is upstream of JNK kinase and JUN, in dedifferentiation via inhibition of adipocyte differentiation. More recently, a role for JUN oncogene amplification on 1p32 in dedifferentiation has been shown. 39,40 Overexpression of JUN results in downregulation of genes involved in adipocytic differentiation, possibly via its direct interaction with CCAAT-enhancer-binding protein beta (C/EBPβ), a transcription factor involved in adipogenesis. 39 Interestingly, JUN amplification has been found in the WDL component (separate from the DDLPS component) of a number of DDLS, suggesting that JUN amplification occurs before dedifferentiation in at least a subset of WDL. 37,40 Inhibition of JUN expression in DDLPS with amplified JUN results in a reduction in proliferation and tumor growth, both in vitro and in vivo, suggesting a key role for JUN upregulation in DDLPS. 40 These in vivo data, however, suggest that JUN amplification is likely not to be sufficient to promote dedifferentiation, as inhibition of JUN expression does not promote adipocyte differentiation in DDLPS tumors. 40
Despite its histology, DDLPS has a less aggressive clinical course than most high-grade pleomorphic sarcomas in adults. The latter tumors typically display both MDM2 and p53 alterations that correlate with a poor prognosis. 41 In contrast, although there are conflicting data regarding the frequency of p53 mutation in DDLPS, 42–44 it is likely mutated in only a minority of these tumors.
Gastrointestinal Stromal Tumor
Clinical Description and Pathology
Gastrointestinal stromal tumors (GISTs) are the most common sarcomas of the gastrointestinal tract. 45 The overall age range at presentation is very broad; however, the majority of tumors are diagnosed in patients older than 50. GISTs occur with equal incidence in both males and females and can occur at any location in the gastrointestinal tract. The most common site is the stomach (50%), followed by the small intestine (25%), large intestine (10%), esophagus (5%), and, rarely, the gallbladder, appendix, or pancreas. GISTs can also arise at sites outside the tubular gastrointestinal tract, including the retroperitoneum, pelvis, mesentery, and omentum, although these extragastrointestinal GISTs account for only about 10% of all GISTs. Clinical presentation may include anemia secondary to gastrointestinal bleeding, early satiety, and intestinal obstruction; however, smaller GISTs are also quite often identified as incidental findings.
The most common metastatic sites for GISTs are intra-abdominal, namely the liver, peritoneum, omentum, and mesentery. GISTs rarely spread to lymph nodes (other than in the pediatric subtype) or to extra-abdominal sites, and when they do, it tends to be late in the course of disease. The most important predictors of metastasis are tumor size and mitotic index, with a size less than 5 cm and a mitotic index less than 5 per 50 high power fields (hpf) conferring a low risk of aggressive behavior and a size greater than 10 cm and/or a mitotic index greater than 10 per 50 hpf conferring a high risk of aggressive behavior. 46 Very occasionally, however, even small GISTs (less than 2 cm) with a low proliferative rate can behave aggressively; thus follow-up is recommended even for lesions with a low relative risk of metastasis.
Based on cytomorphology, GISTs can be divided histologically into three categories: spindle cell, epithelioid, and mixed epithelioid and spindle-cell type. Epithelioid areas are composed of cells growing in sheets or nests, with round nuclei and fairly abundant eosinophilic cytoplasm (Figure 44-2 , A). The spindled cells are typically monomorphic, with vesicular chromatin and palely eosinophilic, almost syncytial cytoplasm, growing in short fascicles (see Figure 44-2, B). Pleomorphism is rare. Additional characteristic features of both spindle-cell and epithelioid GISTs are the presence of perinuclear vacuoles (especially in gastric lesions) and the fibrillary nature of the cytoplasm.
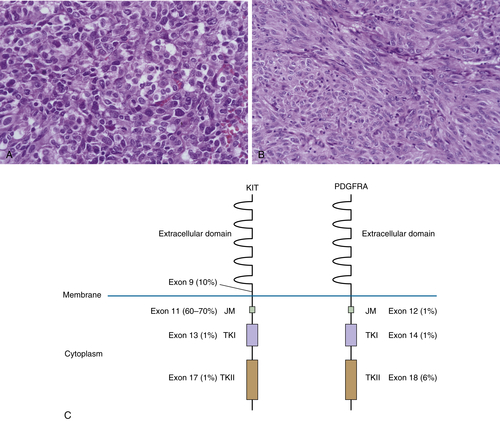
Figure 44-2 Gastrointestinal stromal tumor (GIST) (A) Epithelioid GIST, showing cells with round nuclei and fairly abundant eosinophilic cytoplasm, perinuclear vacuoles, and fibrillary eosinophilic cytoplasm. (B) Spindle-cell GIST, demonstrating the typical monomorphic cells with vesicular chromatin and palely eosinophilic syncytial-appearing cytoplasm, growing in short fascicles. (C) Diagram of c-Kit and PDGFRA, demonstrating localization and frequency of mutations occurring in sporadic GIST. Approximately 5%-10% of GIST lack either c-Kit or PDGFRA mutations. JM, Juxtamembrane domain; TKI, tyrosine kinase domain I; TKII, tyrosine kinase domain II; TM, transmembrane domain.
GISTs frequently stain positively for smooth muscle markers (at least 30% to 40% are positive for smooth muscle actin or caldesmon). In the past, this finding, combined with the eosinophilic quality of the cytoplasm, caused these tumors to often be mistaken for smooth muscle tumors. GISTs were also not uncommonly mistaken for neural tumors because they can exhibit prominent nuclear palisading, mimicking a nerve sheath tumor, and can occasionally (approximately 5%) stain positively for S100 protein, a marker common to neural tumors. However, unlike other mesenchymal tumors of the gastrointestinal tract, the vast majority (95%) of GISTs are positive for the tyrosine kinase receptor c-kit (KIT) (also known as CD117), typically in a diffuse cytoplasmic, dotlike, or membranous pattern. 45 This KIT immunopositivity reflects the effect of activating mutations in KIT (see later discussion) and has greatly improved the reproducibility of GIST diagnosis. More recently, antibodies to DOG-1, a protein highly expressed in GISTs, have been shown to be highly specific, and more sensitive than KIT, in the diagnosis of GIST. 47,48
In the gastrointestinal tract, KIT is also expressed in interstitial cells of Cajal (ICC), gut pacemaker cells exhibiting both smooth muscle and neuronal differentiation ultrastructurally and immunohistochemically, which are important for intestinal peristalsis. 49 Given the similarities between ICC and GISTs both ultrastructurally and immunohistochemically, it is believed that most GISTs show differentiation toward ICC.
Genetics and Molecular Pathogenesis
Approximately 85% to 90% of GISTs harbor activating KIT or PDGFRA mutations 45 (including in familial GIST syndrome, in which patients have germline activating mutations in KIT or PDGFRA, which are inherited in an autosomal dominant fashion). 50 In addition to GISTs, patients with familial GIST syndrome also have ICC hyperplasia, and those with specific mutations in exon 11 of KIT have abnormal skin pigmentation and mastocytosis. The proto-oncogene KIT is a type III receptor tyrosine kinase related to platelet-derived growth factor receptor (PDGFR) and is required for melanogenesis, myelopoiesis, fertility, and ICC and mast-cell development. 51,52 It is a transmembrane protein whose ligand is stem-cell factor (SCF). Binding of SCF to KIT results in KIT activation via autophosphorylation, leading to downstream phosphorylation of key signal transduction proteins and regulation of a number of cell processes, including proliferation, survival, cell adhesion, and differentiation. 53 In GISTs, KIT mutations cluster mainly in four exons: exon 9 (extracellular transmembrane domain), exon 11 (intracellular juxtamembrane domain, exon 13 (initial portion of the kinase domain), and exon 17 (kinase activation loop) (see Figure 44-2, C). Current data suggest that exon 11 mutations are the most frequent (60% to 70% of GISTs). 45 Mutations in exons 13 and 17 are rare.
Platelet-derived growth factor receptor A (PDGFRA) is mutated in approximately 5% of GISTs, resulting in constitutive activation and downstream activation of signal transduction molecules similar to those affected by KIT activating mutations. 54 KIT and PDGFRA mutations are mutually exclusive and the mutations found in PDGFRA map to similar domains on the protein to those found in KIT (see Figure 44-2, C). GISTs with PDGFRA mutations tend to exhibit epithelioid rather than spindle-cell morphology and tend to be more common in the stomach.
A mouse knockin model expressing constitutively active KIT has demonstrated that this constitutive activation is necessary and sufficient for GIST tumorigenesis; 55,56 thus mutation in either KIT or PDGFRA is likely an early step in GIST pathogenesis. However, other recurrent genetic changes have been shown to occur. These include loss of chromosomes 14q, followed by loss of 22, 11p, 9p, or 1p. 45 Loss of 9p, on which the tumor suppressor p16(Ink4a) is located, appears to be associated with a worse prognosis. Gains of chromosome 5p, 20q, 17q, or 8q are also associated with a more aggressive clinical behavior; however, correlation of tumor progression with specific genes in those regions has yet to be demonstrated.
Approximately 5% to 10% of GISTs lack either KIT or PDGFRA mutations (wild-type [WT] GIST). These KIT/PDGFR-intact GISTs tend to occur in the pediatric population, as well as in the setting of specific genetic syndromes, namely, neurofibromatosis type I/Von Recklinghausen’s neurofibromatosis (NFI); Carney triad (CT); and Carney-Stratakis syndrome (CSS). CT, for which a clear genetic inheritance pattern or germline mutation has not been found to date, 57 consists of extra-adrenal paragangliomas, pulmonary chondromas, and multifocal epithelioid GISTs of the stomach. 58 The CSS is characterized by paragangliomas and GISTs and is due to germline mutations in succinate dehydrogenase subunits B, C, or D (SDHB, SDHC, SDHD), inherited in an autosomal dominant fashion. 59,60 SDH is a mitochondrial enzyme complex that functions in the Krebs cycle. Recent data have also identified SDHB or SDHC germline mutations in patients with WT GISTs without a personal or family history to suggest CT or CSS, 61 as well as somatic mutation of SDHA. 62 In addition, loss of SDHB protein expression and decreased SDH complex activity have been demonstrated in WT GISTs in patients without germline or tumoral SDH mutation, further supporting a role for the SDH complex in tumorigenesis in these genetically distinct GISTs. 61 Although the majority of SDHB-deficient GISTs occurs in young patients (hence being designated pediatric-type), comparable lesions with distinctive clinicopathologic features occur more rarely in adults. 63,64
GISTs are not responsive to conventional chemotherapy or radiation, and thus, before the identification of KIT mutations in GISTs and the development of targeted therapy, the outlook for patients with clinically aggressive GISTs was grim. Imatinib mesylate, also known as Gleevec, binds to and inhibits the ATP-binding pocket of both KIT and PDGFRA and is approved to treat patients with metastatic and/or unresectable GISTs. Mutational status appears to be important in predicting response to imatinib, in that patients with exon 11 KIT mutations have a significantly better response than patients with exon 9 KIT mutations or without KIT or PDGFRA mutations. 65 Interestingly, GISTs with either exon 9 KIT mutations or without KIT or PDGFRA mutations are more likely to be responsive to sunitinib, a second-line tyrosine kinase inhibitor approved as therapy for imatinib-resistant GISTs. 66 Given the role of SDH mutation and/or deficiency in a significant subset of WT GISTs, protein expression of SDHB has been proposed as a means to initially triage GISTs into those that likely harbor KIT or PDGFRA mutations (type 1; intact SDHB expression), and those with likely SDH complex deficiency (type 2; absent SDHB expression 67,68 ). This subclassification has important clinical implications, as the type 2 GISTs (WT GISTs) usually do not respond to imatinib. It remains important, however, to consider SDHB expression alone with caution, because of potential difficulties in staining interpretation, and because some tumors classified as type 1 via SDHB positivity (namely, GISTs occurring in the setting of NF1) do not contain KIT or PDGFRA mutations and do not typically respond to imatinib. 69
Unfortunately, a significant number of patients eventually develop secondary resistance to imatinib as well as to sunitinib, primarily via secondary mutations in the KIT kinase domain or through KIT amplification. 66,70,71 KIT has been shown to activate a number of key signal transduction pathways, including the mitogen-activated protein kinase (MAPK), Janus kinase/signal transducers and activators of transcription (JAK/STAT), Src family of tyrosine kinases (SFK), Ras/extracellular regulated kinase (Ras/Raf/Erk), and PI3 kinase signaling pathways. 53,72 Of these pathways, in vitro and in vivo data demonstrate that the PI3 kinase, MAPK, and SFK pathways are activated in GISTs. 72,73 In contrast, phosphorylation of the JAK/STAT kinases STAT1 and STAT3 was not dependent on oncogenic KIT signaling, and STAT5 phosphorylation was not detected in either primary GISTs or GIST cell lines. In addition, inhibition of the JAK/STAT pathway did not inhibit proliferation in GIST cell lines, arguing against a significant role for this pathway in GIST oncogenesis. 74 Elucidation of the comparable importance of downstream targets of KIT in GIST tumorigenesis has important therapeutic implications, particularly for the development of treatment strategies in imatinib-resistant tumors. For example, a secondary mutation in BRAF kinase (a kinase mutated in several cancers, particularly melanoma, papillary thyroid carcinoma, and a subset of colorectal cancers) has been identified in an imatinib-resistant GIST, suggesting a possible role for the potent BRAF inhibitor vemurafenib as second-line therapy. 75 Interestingly, BRAF somatic mutation has also been identified as a rare event in WT GIST. 75
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


