Fig. 40.1
Discovery of imatinib .
Binding of imatinib occurs at the ATP-binding site as indicated by docking studies [28] and X-ray crystallography [29]. It has also been shown from the crystal structure that imatinib inhibits the kinase by binding with high specificity to an inactive form of the kinase. Thus, imatinib is a type 2 kinase inhibitor. Binding to this unusual kinase conformation might contribute to the high selectivity of this compound. Surprisingly, the N-methylpiperazine group (added to increase aqueous solubility and oral bioavailability) was found to have strong interaction with the protein via hydrogen bonds to the backbone carbonyl groups.
In addition to being orally bioavailable, thus more convenient for patients, imatinib lacks some of more serious side effects commonly observed with cytotoxic agents [30]. Imatinib has now been approved in numerous countries for treating the chronic phase of Philadelphia chromosome-positive CML. Through its use in clinic, it was discovered that, in addition to inhibiting BCR-ABL, imatinib effectively inhibited ABL, PDGFR, and SCFR (c-Kit) [31]. In particular, the activity against c-Kit manifested as remarkable clinical activity in c-Kit-positive unresectable or metastatic malignant GISTs, for which it is now also licensed.
In patients with newly diagnosed CML in the chronic phase, daily oral administration of imatinib has demonstrated impressive event-free survival (81 %), freedom from progression (93 %), and overall survival (86 %) rates [32]. However, many patients eventually develop intrinsic or acquired resistance to this first-line therapy [33]. To overcome resistance in CML, a number of second generation inhibitors have been developed [34]. These include imatinib-like inhibitors nilotinib (Tasigna™, Novatis, Approved 2007) (Fig. 40.2) [35], dual SRC/ABL inhibitors dasatinib (Spreycel™, Bristol-Myers Squib, Approved 2006) [36], and bosutinib (Bosulif™, Wyeth, Approved 2012) [37], which have been approved for the treatment of adults in all phases of CML with resistance or intolerance to imatinib. Unfortunately, there are still subsets of mutants remain resistant even with these second-generation compounds, particularly a common BCR-ABLT315I mutant at the gatekeeper position which represents ∼15–20 % of all clinically observed mutants [37–40]. To address this unmet need, several programs targeting development of potent BCR-ABLT315I inhibitors have been established and the progress has been summarized in a number of reviews [41–44]. Ponatinib (AP24534, Iclusig™, Ariad) was developed as a “pan-BCR-ABL” inhibitor which is potently active against BCR-ABLT315I and all other tested BCR-ABL variants and suppresses emergence of resistant mutations in a cell-based screen [45]. In addition, ponatinib was also found to be a multitargeted inhibitor with activity against FLT3, FGFR, VEGFR, c-Kit, PDGFR, Eph, Src family kinases, RET, and Tie2. Ponatinib was approved by FDA for the treatment of T315I positive CML and Philadelphia chromosome positive ALL as well as CML or Philadelphia positive ALL that are resistant or intolerant to prior tyrosine kinase inhibitor therapy in December 2012 through FDA Accelated Approval program. Serious adverse reactions have been reported with ponatinib, with vascular occlusion, heart failure and hepatotoxicity prompting the US FDA to issue boxed warnings. Therefore, clinicians need to consider whether the potential benefits of therapy will outweigh the risks before starting treatment.
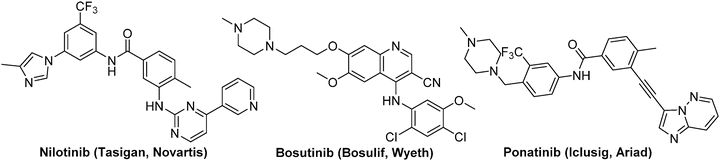
Fig. 40.2
FDA-approved BCR-ABL inhibitors.
40.2.5 EGFR Inhibition
Because of their fundamental role in regulating key cellular functions including cell proliferation, differentiation, metastasis and survival, epidermal growth factor receptor (EGFR) signaling pathways have been a main focus of research for novel targeted anticancer agents [46]. EGFR is a member of the EGFR subfamily known as human epidermal growth factor receptor (HER) family, which contains 4 closely related receptors that include itself (as known as ErbB-1), HER2/neu (as known as ErbB-2), HER3 (as known as ErbB-3) and HER4 (as known as ErbB-4). EGFR is a 170-kDa glycoprotein that is expressed in most human tissues and is highly expressed in many human solid tumors [47].
EGFR is a receptor tyrosine kinase, which has an extracellular domain, a transmembrane domain and an intracellular tyrosine kinase domain. The activation of the EGFR pathway is initiated when an appropriate ligand (e.g., EGF, transforming growth factor-α or TGF-α, heparin-binding EGF, amphiregulin, betacellulin, epiregulin, or neuregulin G2b) [48] binds to the extracellular domain of the inactive single unit of the receptor. This causes receptor dimerization with another identical EGFR receptor to form a homodimer or with a nonidentical receptor from the same EGFR subfamily (e.g., HER2/neu) to give a heterodimer. This dimerization process activates the tyrosine kinase enzyme located in the intracellular domain, and that leads to the transphosphorylation of both intracellular domains. This, in turn, initiates a cascade of phosphorylation events that eventually results in a signal arriving at the nucleus [46]. The magnitude of EGFR signaling is influenced by several cellular mechanisms including receptor mutations, heterodimerization with other members of the HER family, increased expression of autocrine ligands, and alterations in molecules that control receptor signaling output [49].
The work from several research groups has provided strong rationales for EGFR as an anticancer target. EGF was first discovered by Stanley Cohen in the early 1960s [50]. As one of the first growth factors isolated, its discovery opened up a research field that has been crucial to the development of modern anticancer and other medical treatments. A decade later, human EGF4 was isolated [51]. It took another 5 years to demonstrate the link between EGFR and the malignant transformation of cells [52]. An autocrine mechanism was found to be involved in that transformation, involving autostimulation of EGFR in cancer cells by ligands such as EGF and TGF-α, which are produced by the cancer cells themselves. The ligand binding to EGFR activates a cyclic-AMP-independent phosphorylation system through an inherent tyrosine kinase located within the receptor [53]. This up-regulated EGFR-mediated signaling can help move cells into a continuous and uncontrolled stated of division, thus leading to greater numbers of malignant cells and an increase in tumor size. Additional compelling evidence for the role of EGFR in cancer pathogenesis came from reports that many human solid tumors, such as head and neck, lung, and colorectal tumors, express high levels of EGFR, which frequently correlates with poor prognosis [54], in particular, with an increased risk of metastasis and decreased survival. The high expression levels of EGFR can also protect cancer cells from hormone therapies, cytotoxic agents, and radiotherapy [55], thus reducing the effectiveness of these treatments. Furthermore, many tumors that express EGFR also produce one or more EGFR ligands, which further supports the hypothesis that autocrine growth-stimulatory mechanisms are involved in EGFR-mediated tumorigenesis [56]. In addition, it has been shown that EGFR signaling not only increases cell proliferation, but also regulates a range of processes that are essential for tumor progression, including cell motility, cell adhesion, tumor invasion, cell survival, and angiogenesis [57].
40.2.6 The Discovery of Gefitinib (Iressa™)
These strong target validation and disease association results have prompted the research community to pursue small molecule EGFR-TK (tyrosine kinase) inhibitors as potential anticancer therapeutics. These small molecule inhibitors can compete with ATP binding to the TK domain of the receptor, which inhibits TK activity and subsequently block signal transduction from the EGFR. Gefitinib (Iressa™), an 4,6,7-trisubstituted-4-quinazolinamine discovered by AstraZeneca in the mid- to late-1990s, was one of the first agents in this new family [49].
The discovery of gefitinib started from a phenylaminolquinazoline (compound 3A, Fig. 40.3), which was discovered from the SAR study of the 4-anilinoquinazoline class as EGFR-TK inhibitors [58, 59]. This compound was found to be a potent inhibitor of EGFR-TK (IC50 = 5 nM) and EGF-stimulated human cell growth (IC50 = 50 nM (KB oral carcinoma cells)]. However, compound 3A had a short half-life (approximately 1 h) and appeared to undergo rapid oxidative metabolism. Substitution of the methyl group with chlorine and introduction of fluorine at the para-position of the aniline gave compound 3B, which was resistant to the oxidative metabolism. Although this compound showed a slight loss of potency in vitro (EGFR-TK, IC50 = 9 nM; EGF stimulated KB cell growth, IC50 = 80 nM) compared to compound 3A, it had improved efficacy in in vivo models via oral dosing and better pharmacokinetic (PK) properties (e.g., reduced clearance, longer half-life—approximately 3 h). The approach aimed at further improving in vivo activity, which eventually proved successful, involved modification of the alkyl group of the methoxy side chain. Introduction of a basic group into the alkoxy side chain resulted in improved physical chemical properties such as solubility and further improved PK parameters. After profiling a series of compounds with different aminoalkoxy side chains, gefitinib (ZD1839) (Fig. 40.3) was chosen as a development candidate based on its excellent in vitro and in vivo profiles. Although gefitinib was not the most potent compound synthesized, it achieved high and sustained blood levels over a 24-h period and had good oral bioavailability in preclinical species [60]. Gefitinib is selective against structurally closely related ErbB2, the receptors of VEGF, FMS like tyrosine kinase (FLT1; VEGFR1), kinase insert domain-containing receptor tyrosine kinase (KDR; VEGFR-2), and a number of serine/threonine kinases [61]. Gefitinib inhibits the growth of a broad range of human solid tumor xenografts in a dose-dependent manner with marked regressions seen in some tumors. Treatment for up to 4 months in nude mice was well tolerated. Gefitinib has demonstrated a long half-life in humans compatible with once-daily oral dosing. The FDA approved gefitinib as a second-line treatment for non-small-cell lung cancer in May 2003.
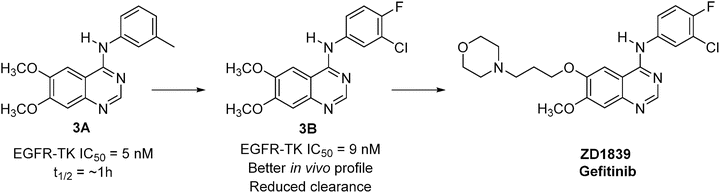
Fig. 40.3
Discovery of gefitinib.
40.2.7 The Discovery of Afetinib (Gilotrif™)
As exemplified by etitinib and erlotinib (Tarceva™, OSI), the first-generation small molecule EGFR tyrosine kinase inhibitors were shown to be effective against lung tumor cells harboring mutations in the kinase domain of EGFR, most commonly small in-frame deletions in exon 19 or the L858R missense mutation in exon 21 [62]. However, despite the initial response, patients almost invariably develop resistance to these inhibitors and relapse after several months [63]. A secondary mutation, T790M, in exon 20 of the EGFR kinase domain is account for about half of cases with acquired resistance to first-generation EGFR inhibitors [64, 65]. T790M EGFR exhibits elevated enzymatic and transforming activity, both alone and in combination with primary alterations in exon 19 or 21 [66–69], indicating a need for increased therapeutic efficacy of the next generation of EGFR inhibitors. Ideally, these new small molecule tyrosine kinase inhibitors should be more broadly active against ErbB receptor tyrosine kinases, yet retain the exquisite overall selectivity of first-generation EGFR TKIs within the human kinome that has afforded an acceptable drug safety and tolerability profile.
Irreversible inhibitors that covalently modify EGFR and/or HER2 exhibited increased efficacy against mutants resistant to gefitinib and erlotinib in cell-based assays [64, 69–73]. One such inhibitor, afetinib (BIBW2992) (Fig. 40.4), is a new irreversible dual EGFR/HER2 inhibitor derived from the anilino-quinazoline chemical series that was designed to covalently bind to Cys 773 of EGFR and Cys 805 of HER2 via a Michael addition [74]. Afetinib was found to potently suppress the kinase activity of wild-type and activated EGFR and HER2 mutants, including the most common mutations, L858R and deletion-19, and the exon 20 gatekeeper T790M mutations, albeit at lower potency [74, 75]. It has received regulatory approval for use as a treatment for non-small-cell lung cancer in 2013 although there is emerging evidence to support its use in other cancers such as breast cancer [76].
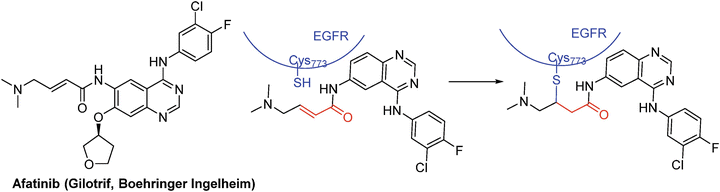
Fig. 40.4
Afatinib and its inhibitory mechanism.
40.2.8 The Discovery of Lapatinib (Tykerb™)
Among the four members (EGFR, HER2, HER3, and HER4) of the EGFR family, only HER2 is unknown to bind ligands [77]. However, HER2 is the preferred heterodimerization partner for EGFR, HER3, and HER4. Ligand binding to the extracellular domain of members of this family causes receptor homodimerization or heterodimerization and tyrosine kinase activation. The resultant receptor autophosphorylation leads to the activation of various signaling cascades that are involved in cell proliferation and survival [78].
EGFR and HER2 have been shown to promote the growth and survival of various types of epithelial malignancies. EGFR is overexpressed in up to 30 % of human breast cancers [79], and HER2 is amplified and overexpressed in up to 20–25 % of primary human breast cancers [80]. Importantly, overexpression of HER2 is associated with aggressive disease and shortened disease-free survival and overall survival [81]. Abundant evidence suggests that EGFR and HER2 work in synergy to produce oncogenic effects [82]. In particular, HER2 dimerization with EGFR increases the EGF binding affinity to EGFR [83], and the binding of EGF to EGFR subsequently increases the activation of HER2 [84]. It was therefore hypothesized that agents that inhibit both EGFR and HER2 might be more effective at inhibiting the activation of the PI3K-Akt and Ras-Raf-mitogen-activated protein kinase signaling cascades and could have more effective anticancer activity than those that target either kinase alone. Efforts on developing such molecules led to the discovery of lapatinib (Tykerb™, GlaxoSmithKline) [85].
The discovery of lapatinib (originally known as GW572016) started with the screen hit 4557 W (Fig. 40.5) [86], which was a potent inhibitor of both HER2 (IC50 = 0.079 μM) and EGFR (IC50 = 0.020 μM). Initial SAR study demonstrated that large anilino substitutions were needed to retain both EGFR and HER2 inhibition activity. The most potent dual enzyme inhibition could be obtained when the aryl group was the substituted benzyloxyaniline group, and the optimal substitution pattern involved a para-benzyloxy group. Other notable SAR include that although chlorine at 3-position afforded insignificant increases in enzyme potency, it improved cellular activity. Fluorine at 2′-position yielded compounds with greater potency in cellular assays. Larger groups at 3-position or 2′-position generally diminished activity.
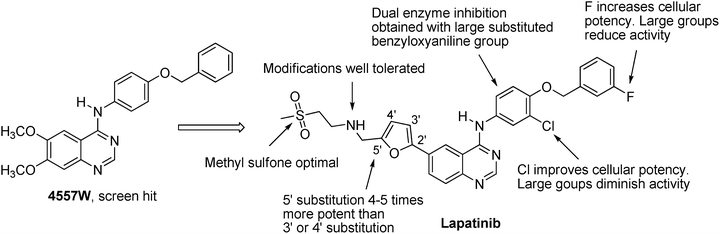
Fig. 40.5
Discovery of lapatinib.
For the left-hand side capping group, the simple methyl sulfone group appeared to be optimal. Study on the regiochemistry of the furan ring showed that the 5′ substituted analog was 4–5 times more potent against the tumor cell lines than the 3′ or 4′ regioisomer. In addition, a diverse set of amine substitutions was tolerated, presumably due to the binding mode of these inhibitors. However, the extra synthetic steps and increased molecular weight were deemed nonbeneficial for optimizing physical chemical properties.
The medicinal chemistry optimization ultimately led to the discovery of lapatinib, which possessed the desired enzyme potency, cellular activity in a panel of tumor cell lines, and excellent selectivity [87]. In March 2007, FDA approved lapatinib for treating breast cancer.
40.2.9 VEGFR Inhibition: The Discovery of Pazopanib (Votrient™) and Axitinib (Inlyta™)
Angiogenesis plays a critical role in the growth and metastasis of solid and hematologic malignancies [88]. It is considered as the rate-limiting step in tumor development because without blood supply, a solid tumor is limited to a maximum size of 1–2 mm [89]. This complex and highly regulated process involves numerous different cell types and mediators.
VEGF is the growth factor most often associated with angiogenesis [90]. Oxygen-deficient cells use VEGF in key signaling pathways to promote the growth of new blood vessels. VEGF interacts with VEGFRs on endothelial cell surfaces, directing them to build new blood supplies [91]. The cells respond to this message by producing specialized protease enzymes to penetrate the basal lamina, so that they can migrate to oxygen-deficient regions. Once there, the cells replicate and form into tubes, thus creating new capillary pathways. Elevate VEGF levels have been correlated with increased microvessel counts and poor prognosis in many cancer types [92].
As a principal subfamily of RTKs, VEGFRs are specifically expressed in vascular endothelial cells. They include FMS like tyrosine kinase-1 (FLT-1; VEGFR-1), kinase insert domain-containing receptor tyrosine kinase (KDR; VEGFR-2), and FLT-4 (VEGFR-3) [93]. Activation of VEGFR family RTKs and, in particular, VEGFR-2 by VEGF plays a primary role in tumor angiogenesis. VEGF mediated VEGFR-2 signaling induces a series of endothelial responses such as proliferation, migration, and survival and ultimately leads to new vessel formation and stabilization [94].
The therapeutic hypothesis that inhibiting the VEGF/VEGFR pathway can interrupt the angiogenesis process and subsequent tumor growth was first clinically validated by Bevacizumab (Avastin™; Genentech), a monoclonal antibody to VEGF, which is approved by FDA for the treatment of both metastatic colorectal cancer in combination of 5-fluorouracil [95], and for non-small-molecule lung cancer in combination with carboplatin and paclitaxel [96]. Efforts on discovering small molecule inhibitors of VEGFRs [97] led to the FDA approval of pazopanib (Votrient™, GlaxoSmithKline), N4-(2,3-dimethyl-2H-indazol-9-yl)-N4-methyl-N2-(4 methyl-3sulfona midophenyl)-2,4-pyrimidinediamine, as an oral treatment for renal cell carcinoma (RCC).
The discovery of pazopanib started from a screening for agents active against VEGFR-2 [98]. Initial screening hit compound 6A (Fig. 40.6), which inhibited VEGFR-2 with an IC50 of ∼400 nM, was optimized to compound 6B using a homology model of the kinase domain as the crystal structure was not known at that time. Compound 6B inhibited VEGFR-2 with an IC50 of 6.3 nM, almost a 100-fold potency improvement compared to 6A. In cellular assays, compound 6B inhibited VEGF induced proliferation of human umbilical vein endothelial cells (v-HUVEC) with an IC50 of 0.54 μM. It also had an IC50 of 3.5 μM against basic fibroblast growth factor (b-FGF). However, the pharmacokinetic profile of this compound was poor (moderate to high clearances, 28–83 mL/min/kg, and low oral bioavailability, <11 %) in mice. Incorporation of a 3-methylindazole moiety into this series yielded indazolylpyrimidine (compound 6C) with desired cellular potency, and selectivity in VEGF and bFGF driven HUVEC, with IC50’s of 0.13 and 13 μM, respectively. 6C had moderate plasma clearance (40 mL/min/kg) and oral bioavailability (28 %) in rats. Introducing a methyl group into the C-4 amino nitrogen resulted in compound 6D, which showed an improved PK profile with lower clearance (10 mL/min/kg) and higher oral bioavailability (65 %) while providing comparable enzymatic potency to its non-alkylated analog 6C. Although compound 6D possessed both good in vitro potency and cross-species PK profiles, significant inhibition (IC50 < 10 μM) was observed against a number of cytochrome P450 isozymes possibly due to the binding of the nitrogen on the indazole to the heme iron of the cytochrome P450 enzymes. Compound 6E resulting from the addition of a methyl group at the 2-position of the indazole ring, which increases steric hindrance of the heterocycle ring, had an improved profile against the CYP2C19, CYP2D6, and CYP3A4 isozymes as well as improved cellular efficacy against v-HUVEC cells. With the preferred right-hand side moiety identified, a second aniline survey at the C-2 position of the pyrimidine resulted in the discovery of compound 6F (pazopanib), which possesses a desirable combination of excellent in vitro potency, selectivity, cellular activities, PK, and developability properties. Pazopanib was potent against all human VEGFR receptors with a respective IC50 of 10, 30, and 47 nM for VEGFR-1, -2, and -3. Significant activity was also seen against the closely related tyrosine receptor kinases PDGFRα, PDGFRβ, c-Kit, FGF-R1, and c-fms with IC50’s of 84, 74, 140, and 146 nM, respectively. In cellular assays, pazopanib had a desirable combination of cellular potency and selectivity in VEGF and bFGF driven HUVEC, with IC50’s of 0.021 and 0.72 μM, respectively. It also potently inhibited VEGF induced phosphorylation of VEGFR-2 in HUVEC cells with an IC50 of 8 nM. Pazopanib had good PK parameters including low clearances and good oral bioavailability in preclinical species. The cytochrome P450 profile was also significantly improved with IC50 > 10 μM against the isozymes tested, with the exception of CYP2C9 (IC50 = 7.9 μM). On the basis of its favorable PK profiles and in vivo efficacies, the mono-HCl salt of compound 6F (pazopanib) was progressed into clinical development [98] and approved by the FDA for the treatment of renal cell carcinoma in October 2009.
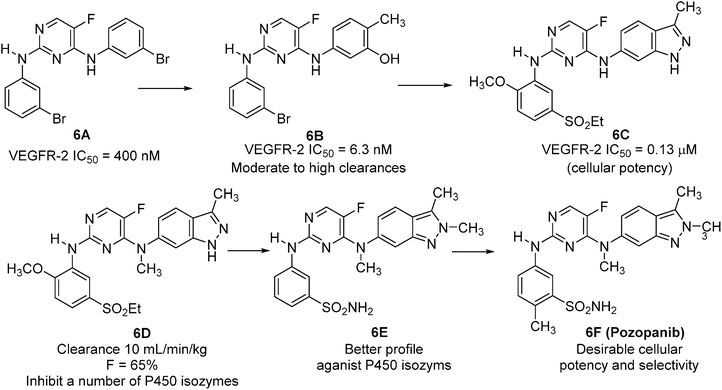
Fig. 40.6
Discovery of pozopanib.
Axitinib (AG013736, Inlyta™, Pfizer) (Fig. 40.7), approved by FDA in January 2012, is another ATP-competitive VEGFR inhibitors for patients with metastatic renal cell carcinoma (mRCC) after failure of one prior systemic therapy [99]. This small molecule indazole derivative is a potent, orally bioavailable multitargeted tyrosine kinase receptor inhibitor, which selectively inhibits vascular endothelial growth factor receptors (VEGFR)-1, -2, and -3 at sub-nanomolar concentrations, in vitro [100]. The structure-based drug design of axitinib allows strategic optimization of critical binding elements, with the tight fit of axitinib into the deep pocket conformation of the kinase domain of VEGFRs resulting in high potency and selectivity (Fig. 40.7). In vitro, axitinib inhibits VEGFR-1, -2, and -3 autophosphorylation at picomolar concentrations. Axitinib is tenfold more potent for the VEGF family receptors than for RTKs of other family receptors [100]. Additionally, axitinib did not significantly inhibit other receptor kinases including colony-stimulating factor (CSF)-1R, fms-like tyrosine kinase (Flt)-3, fibroblast growth factor receptor (FGFR)-1, ret proto-oncogene (RET), epidermal growth factor receptor (EGFR), and met proto-oncogene encoding hepatocyte growth factor (c-Met) [100].
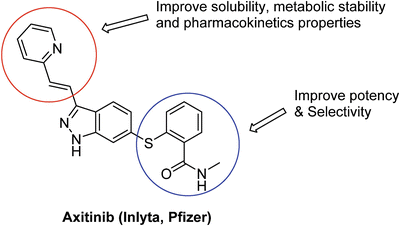
Fig. 40.7
Drug design strategy for axitinib.
Axitinib , orally administrated at a dose 2–10 mg twice daily, has been shown to reduce vascular permeability, tumor vascularization and tumor volume, and has demonstrated antitumor activity as a single agent in patients with cytokine- and/or sorafenib-refractory mRCC. The activity range of axitinib is the highest compared with other drugs currently approved for use in mRCC. Axitinib also has a favorable and non-cumulative tolerability profile associated with manageable adverse effects, which are generally mild to moderate in severity [101]. Based on the promising results from clinical trials, FDA approved axitinib for the treatment of mRCC after failure of one prior systemic therapy in 2012.
40.2.10 Multitargeted Tyrosine Kinase Inhibition
In search for more effective anticancer therapeutics, the multitargeted approach has emerged as a new paradigm [102]. Malignant tumors are complex tissues involving interplay between cancer cells and surrounding or supportive cells, thus a multitargeted therapeutic approach that affects supportive tissues and cancer cells may be of clinical benefit. Such agents can simultaneously target the tumor and surrounding or supportive cells and thereby interact with the complex multimolecular lesions that drive tumor growth and survival. A key advantage of the multitargeted approach is that it potentially reduces drug resistance. Due to mutations [72], overexpression of key components of signaling pathways [103], drug-efflux systems and/or signaling bypass [104], tumors will likely develop resistance to single-targeted kinase inhibitors rapidly [105]. This drug resistance will be less likely to arise with multitargeted agents. In addition to multitargeted agents, the use of a combination of multiple kinase inhibitors has been investigated, which could offer similar benefits as multitargeted agents. In this section, we use sunitinib and dasatinib as examples to illustrate the success of multitargeted kinase inhibitor agents.
40.2.11 The Discovery of Sunitinib (Sutent™)
Growth factors including VEGFs and PDGFs are important pro-angiogenic factors. Their activity is mediated by binding to specific cell-surface receptor tyrosine kinases. There are four PDGF ligands (PDGFA to PDGFD), which bind to the receptor tyrosine kinases PDGFRα and PDGFRβ. PDGFB and PDGFRβ have been suggested to have crucial roles in tumor-vessel stability by recruiting pericytes to newly formed vessels [106]. The multitargeted tyrosine kinase inhibitor sunitinib (Sutent™, Pfizer) was designed to inhibit both VEGFRs and PDGFRs. Sunitinib, which demonstrated activity in RCC and other types of cancers, has been approved by the FDA for the treatment of kidney cancer and GISTs in January 2006.
Sunitinib (Sutent™, formerly SU11248, named from Schlessinger and Ullrich who created Sugen, a biotech company later acquired by Pharmacia then Pfizer) [107] is an orally active oxindol that has antiangiogenic and antitumor activities. It inhibits VEGFR-1, VEGFR-2, VEGFR-3, PDGFRα, PDGFRβ, KIT, FLT3, glial cell line-derived neurotrophic factor (GDNF) receptor, and colony stimulating factor 1 (CSF1) receptor (CSF1R) [108]. Sugen initially discovered SU5416 (Fig. 40.8) as a potent and selective VEGFR inhibitor [109]. Both in vivo and in vitro studies demonstrated its antiangiogenic potential [110]. Unfortunately, SU5416 failed in clinical trials due to its limited solubility and high toxicity. SU6668, a close analog of SU5416, was identified as a potent and selective PDGFRβ inhibitor [111, 112]. However, this compound had inadequate PK properties for clinical development. To broaden the kinase activity spectrum of this scaffold and to optimize PK properties, diversification at the C-4′ position on the pyrrole ring of SU5416 had been explored. It was found that modifications at the C-4′ position could lead to compounds with different kinase inhibition profiles for VEGFR2 and PDGFRβ. The cocrystal structure of SU6668 with the catalytic domain of fibroblast growth factor receptor 1 (FGFR1) kinase [111], which has high sequence similarity to the ATP-binding pocket of VEGFR-2, revealed that the substitution at the C-4′ position on the pyrrole ring was positioned close to the opening of the binding pocket and could be exposed to solvent. Thus, substitution at this position might serve as a handle for improving physical chemical properties of the core structure. Optimization of this region resulted in the discovery of sunitinib (SU11248), a more potent inhibitor of VEGFRs and PDGFRs. Comparing to SU6668, SU11248 was about 30-fold more potent against VEGFR-2 and PDGFRβ in biochemical assays, over ten-fold more potent in cellular kinase assays, and significantly more soluble under neutral (20-fold) and acidic (>500-fold) conditions [112]. Additional kinase selectivity study revealed that SU11248 was also a good inhibitor of KIT and FLT-3 [108, 113]. Furthermore, SU11248 had excellent oral bioavailability, was highly efficacious in a number of preclinical tumor models, and was well tolerated at efficacious doses [108]. Based on the satisfactory results from clinical trials, sunitinib was approved by the FDA for the treatment of RCC/GISTs in January 2006.
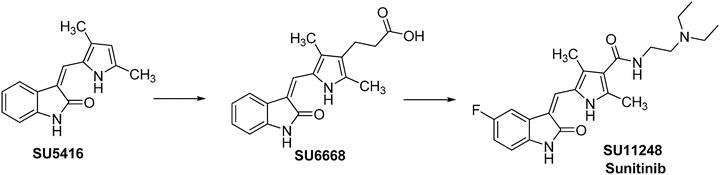
Fig. 40.8
Discovery of sunitinib.
40.2.12 The Discovery of Dasatinib (Sprycel™)
Dasatinib (BMS-354825, Sprycel™; Bristol Myers Squibb) is a potent, second-generation, multitarget kinase inhibitor of BCR-ABL and the Src (sarcoma) family of kinases [114]. In addition to inhibiting BCR-ABL and Src kinases such as Fyn (Proto-oncogene tyrosine-protein kinase), Yes (Proto-oncogene tyrosine-protein kinase), Src, and Lyk (tyrosine-protein kinase), dasatinib was found to inhibit ephrin receptor A2 (EphA2), PDGFRs, c-Kit, mitogen-activated protein kinases, and discoidin domain receptor 1 (DDR1) [115].
Src proto-oncogene is believed to play a role in the development of several human cancers, contributing to cellular proliferation, adhesion, invasion, and motility [116]. Src-family kinases such as Src and LYN (V-yes-1 Yamaguchi sarcoma viral related oncogene homolog) modulate signal transduction by phosphorylating tyrosine residues of molecules such as EGFRs, PDGFRs, fibroblast growth factor receptors (FGFRs), and VEGFRs. Src kinases have also been found to have oncogenic activities in cancer cell lines from tumors involving the colon, breast, pancreas, lung, and brain [117]. Thus, it was hypothesized that blocking signaling through the inhibition of the kinase activity of Src would be an effective means of modulating aberrant pathways that drive oncologic transformation of cells.
The discovery of dasatinib started from 2-acylamino-5-carboxamidothiazoles 9A (Fig. 40.9), a Lck (lymphocyte-specific protein tyrosine kinase) kinase inhibitor [118]. Replacing the 2-acyl functionality with a variety of heterocycles led to the identification of compound 9B [119], which was a potent inhibitor of multiple Src-family members including Lck, Fyn, Src, and Hck (hemopoietic cell kinase). In order to improve cellular activity of this series, a number of 9B analogs were synthesized. These efforts resulted in the discovery of the piperazinyl ethanol 9C (dasatinib), which has broad spectrum antiproliferative activity, favorable circulating plasma levels following oral dosing, long half-life, and favorable in vivo efficacy [120].
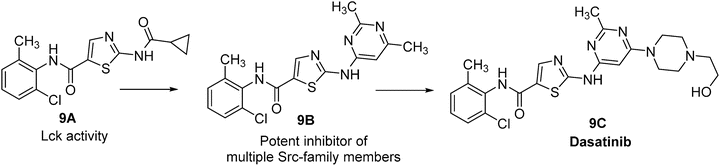
Fig. 40.9
Discovery of dasatinib.
In addition to inhibiting multiple Src-family kinases including Lck, Fyn, Src, and Hck, dasatinib is a BCR-ABL inhibitor that blocks both the active and inactive conformations of ABL. Dasatinib is active against wild-type and almost all BCR-ABL mutant cell lines that are resistant to imatinib, except those carrying the T315I mutation [36]. The activity of dasatinib against ABL kinase mutations is likely due to the fact that it does not require interaction with some of the residues involved in those mutations. Dasatinib is also 325-fold more potent than imatinib against native BCR-ABL in in vivo studies [121] and is slightly more potent against Src family kinases (IC50 = 0.5 nM) compared to ABL (IC50 = 1 nM) [120]. It also inhibits c-KIT, PDGFR and EphA2 at nanomolar concentrations [115]. Potent Src inhibitory activity of dasatinib in CML progenitors involves BCR-ABL-dependent and BCR-ABL-independent Src activity [122]. Dasatinib also inhibits downstream signaling pathways in CML progenitors, as well as P-mitogen-activated protein kinase (MAPK), P-protein kinase B (Akt), and P-signal transducer and activator of transcription 5 (STAT5) levels in CML progenitors in the absence of growth factors. In addition, dasatinib significantly suppresses CML colony-forming cells and long-term culture-initiating cells [122]. In June 2006, the FDA approved dasatinib for the treatment of chronic phase, accelerated phase, or blastic phase CML, resistant or intolerant to imatinib, and for the treatment of Philadelphia chromosome-positive acute lymphoid leukemia that was resistant or intolerant to prior therapy.
40.2.13 Other Potential Kinase Targets
Encouraged by the rapid progress and tremendous success achieved in the fields of cancer molecular biology and kinase drug discovery to date, the academic research community and the pharmaceutical and biotech industry are targeting many other kinases for achieving potential clinical benefits. For examples, inhibitors of integrin-linked kinase (ILK), a focal adhesion serine–threonine protein kinase, are being developed for treating anaplastic thyroid cancer [123]. Focal adhesion kinase (FAK) has been shown to be an important mediator of cell growth, cell proliferation, cell survival, and cell migration [124]. Cell division cycle 7-related protein kinase (Cdc7 ) is a conserved serine–threonine kinase, which has been shown to be required for the initiation of DNA replication [125]. Other areas of research include the hepatocyte growth factor receptor (MET) [126], and the Ras/ERK1-2 (extracellular signal-regulated kinase 1-2) [127], Akt [128] and STAT (signal transducer and activator of transcription) [129] pathways. In this section, we describe the discovery of PD0325901, ARRY-142886, and trametinib (GSK1120212), three inhibitors of mitogen-activated protein kinase kinases (MEK), to highlight recent progress in this new area.
40.2.14 MEK Inhibition
The signaling pathway comprising mitogen-activated protein kinase (MAPK) kinases (MEK) /extracellular signal-regulated kinases (ERK) is activated in most human tumors [130]. Activation of the ERK pathway involves the guanosine triphosphate (GTP)-loading of Ras at the plasma membrane, and the sequential activation of a series of protein kinases. Initially, activated Ras recruits the Raf family of kinases such as Raf-1 to the plasma membrane, where it is then able to phosphorylate and activate MEK1/2 by serine phosphorylation. MEK1 and 2 then phosphorylate and activate their only currently known substrates, ERK1 and 2. When activated, ERK1 and 2 phosphorylate various downstream substrates involved in a multitude of cellular responses such as cell proliferation, cell differentiation, cell survival and cell motility [131].
Because the Raf-MEK-ERK kinase signaling module is activated as a consequence of several gene amplifications and genetic mutations identified in human cancer, it is an attractive target for small-molecule intervention [127]. Selective MEK1/2 inhibitors could be valuable tools to evaluate the role of this signaling axis because MEK1 and 2 have extremely unique characteristics among the components of the ERK pathway. Although Raf-1 is the major activator of MEK1/2, these kinases are activated also by several other kinases such as Mos, A-Raf, and B-Raf. However, no substrates for MEK1/2 have been identified other than ERK1 and ERK2. In addition, MEK1 and 2 are dual-specificity kinases that phosphorylate both tyrosine and threonine residues and can integrate many mitogenic signaling pathways into the ERK pathway. All these very prominent features including unusually restricted and unique substrate specificities and the integrating role of mitogenic signaling pathways highlight MEK1/2 as excellent targets for developing inhibitors against the ERK pathway [127]. Currently, a number of potent and selective MEK1/2 inhibitors have entered clinical development [132]. Most of them are ATP noncompetitive which means they do not directly compete for the ATP-binding site. Rather they bind to a unique allosteric site adjacent to the ATP site. This makes the noncompetitive MEK inhibitors highly specific [133].
From a screening campaign using an in vitro cascade assay, PD98059 (Fig. 40.10) was identified as the first pharmacological inhibitor of MEK1/2 [134]. Meanwhile, U0126 (Fig. 40.10) was discovered by screening a total of 40,000 compounds in a cell-based reporter assay [135]. Because of their poor PK and developability properties, these two early MEK1/2 inhibitors were not progressed into clinical development. Nevertheless, these two compounds were widely used as in vitro tools to elucidate the role of the ERK pathway in a variety of biological processes. The results from these studies have strengthened the hypothesis that the ERK pathway represents an attractive target for developing anticancer drugs.
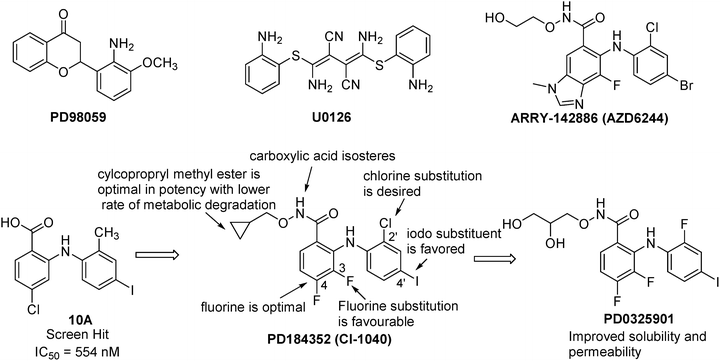
Fig. 40.10
Discovery of MEK inhibitors.
The discovery of PD0325901 began with the high throughput screening hit 10A (Fig. 40.10), a potent inhibitor of MEK with an IC50 of 554 nM [136]. However, this compound had poor cellular potency likely due to poor cell membrane permeability resulting from the carboxylic acid moiety. In order to improve cellular potency, carboxylic acid isosteres such as benzohydroxamic acid esters were investigated. A variety of small alkyl and cycloalkylmethyl esters showed comparable potency with cyclopropyl methyl ester being the best. SAR studies also showed that the iodo substituent was favored at the 4′-position and a chloro substituent was desired at the 2′-position. Additionally, a halo substituent at the 4-position, in particular, a fluorine was optimal as the MEK1 X-ray cocrystal structure revealed that the fluorine had a dipolar interaction with the amide backbone [137]. Substitution of fluorine at 3-position gave moderate potency enhancement. These SAR studies resulted in the discovery of PD184352 (CI-1040), which demonstrated in vivo activity in a mouse tumor model following oral administration [138]. This compound was advanced into Phase II clinical trial, but failed for inadequate efficacy likely due to insufficient systemic exposure. The solubility of PD184352 is less than 1 μg/mL in pH 6.5 phosphate buffer, suggesting that low in vivo exposure is at least partly due to solubility limitations. In order to improve aqueous solubility of PD184352, replacing the cyclopropylmethoxy group with a (R)-dihydroxypropoxy group and the 2′-chloro substituent with a 2′-fluoro substituent led to the discovery of PD0325901, a second-generation MEK inhibitor. This compound has markedly improved potency, oral bioavailability, and half-life. Excellent anticancer activity has been demonstrated for PD0325901 against a broad spectrum of human tumor xenografts [136]. In an open-label, phase II study, patients with progressive, recurrent, or advanced NSCLC were treated with 15 mg PD-0325901 twice daily [139]. There were no objective responses during the trial period. Due to a lack of responses coupled with the safety issues, the trial was closed after the first stage [132].
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


