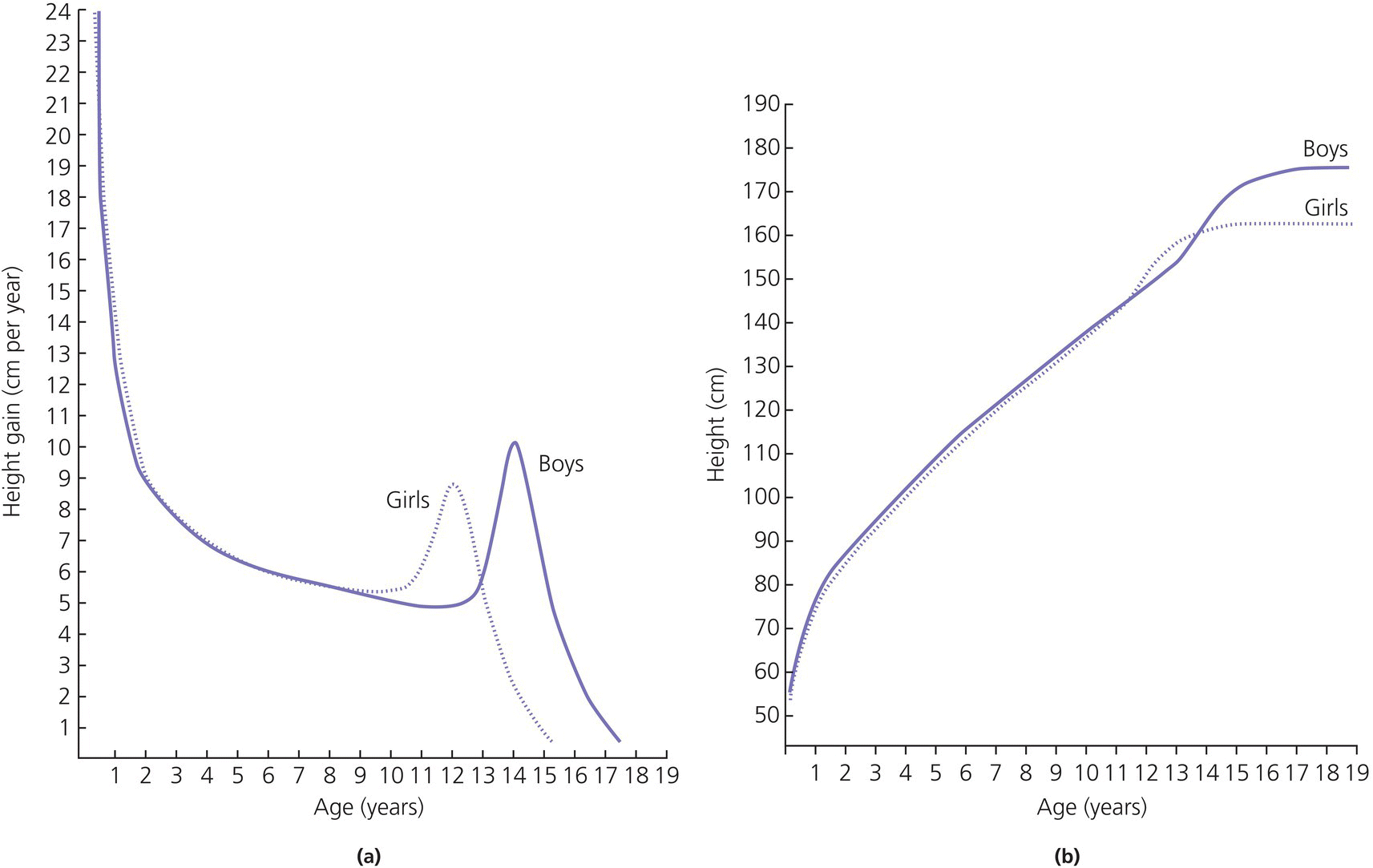
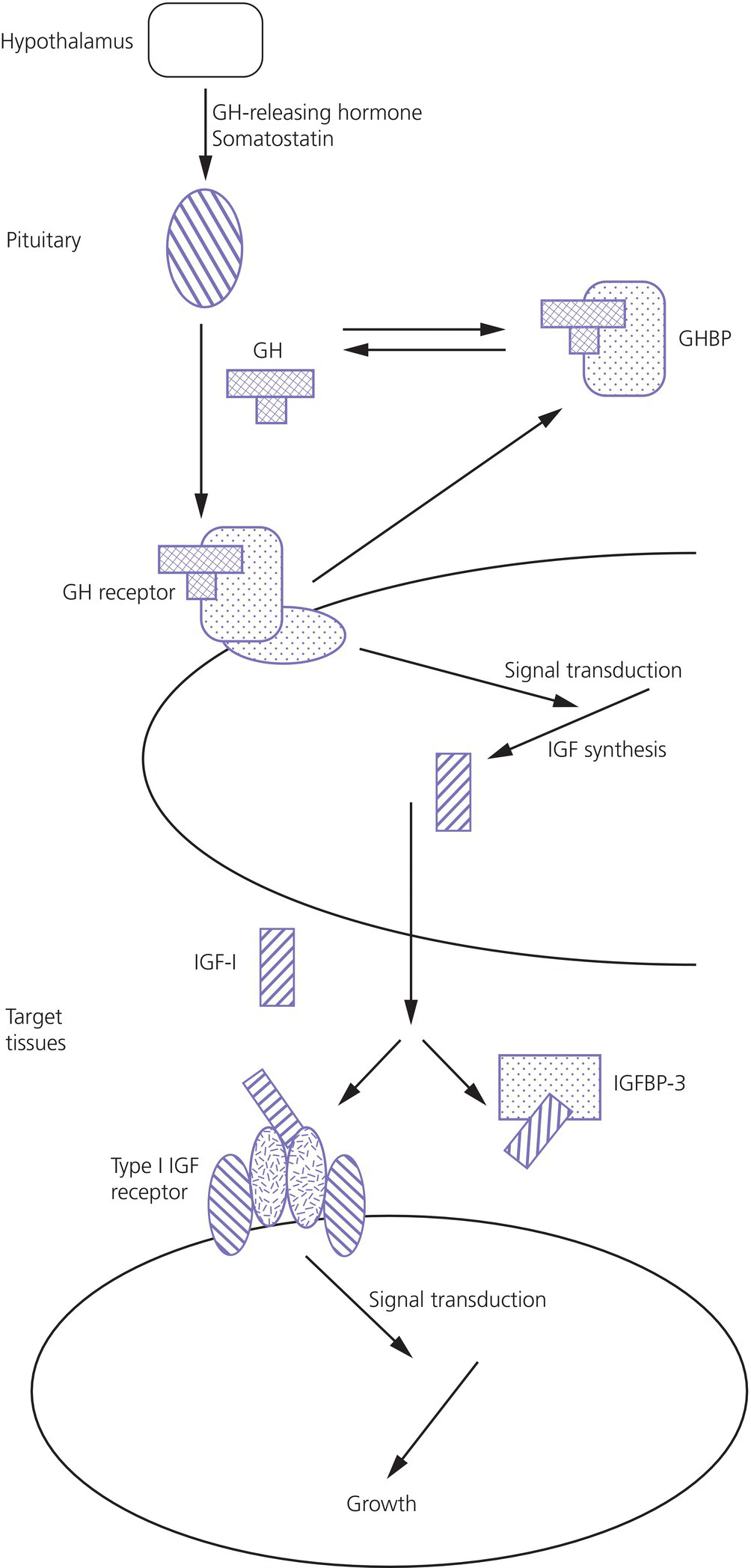
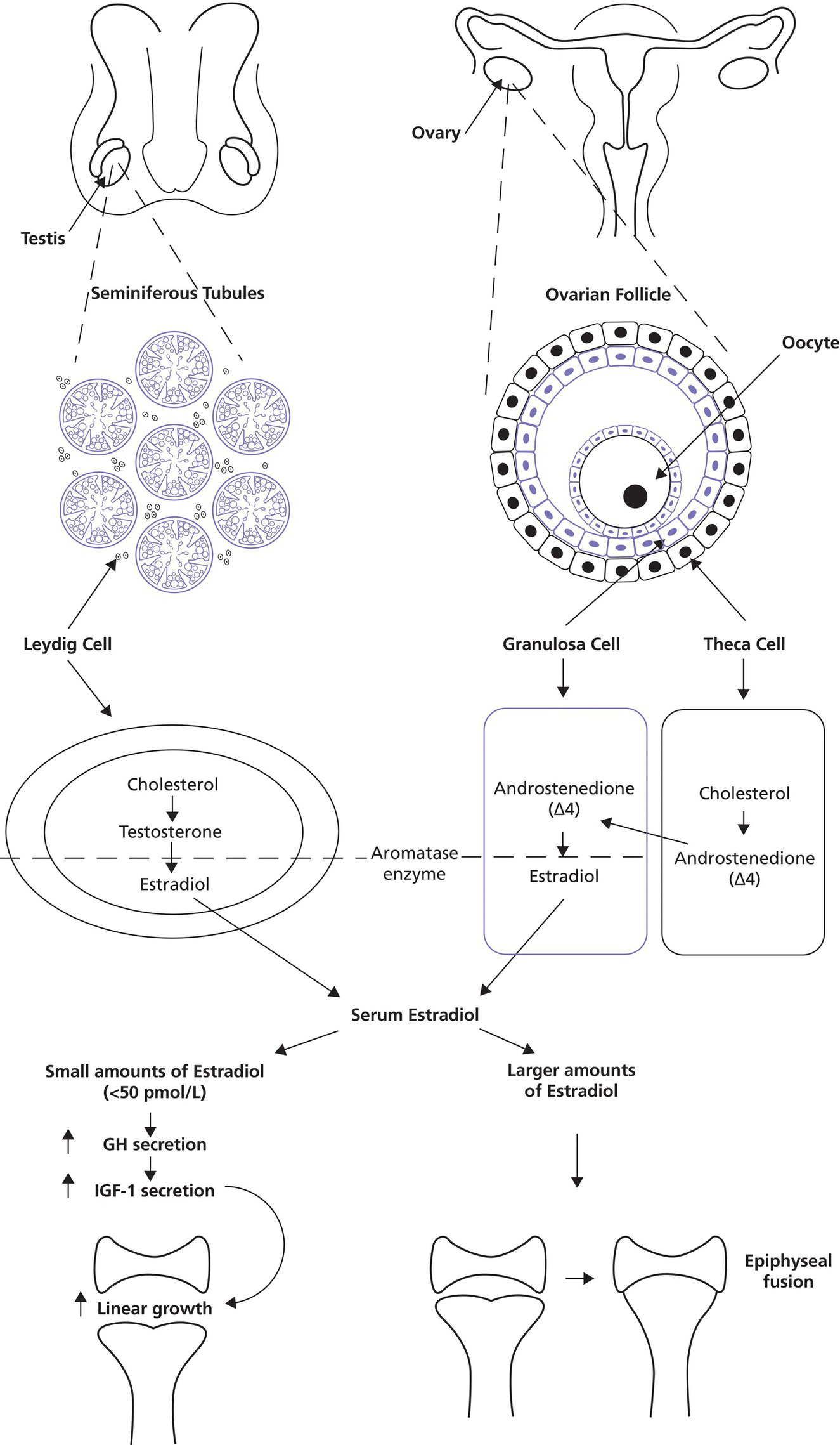
3 The term ‘auxology’ (Greek root, αὐξάνειν, auxanein, to grow) is used to describe the study of human growth using repeated measurements of the same individual over successive time periods. It was Professor James Mourilyan Tanner (1920–2010), who in the 1970s established auxology as a scientific discipline and introduced its routine use as an essential part of clinical growth assessment. Short stature can be defined both in terms of auxology and perception. In auxological terms, short stature refers to height which is less than two standard deviations below the mean for the population concerned; this corresponds to height below the 2nd or 3rd centile, depending on which growth charts are used. In terms of perception, short stature can be defined as small size sufficient to cause physical, psychological, or social concerns in the child and family. Short stature should be distinguished from failure to thrive or weight faltering, a term usually applied to infants and pre‐school children which denotes failure to gain weight at an appropriate rate so that the child looks thin; and growth failure – failure to maintain a height velocity which is appropriate for both age and maturity. In most patients, short stature is a variation of normal physiology rather than a pathological process. The challenge to the paediatrician is to identify the relatively few children with pathology without subjecting essentially normal children to unnecessary investigation. Human linear growth can be divided into three phases – infancy, childhood. and puberty as described by Karlberg (Karlberg 1989 – see Figure 3.1). These are not distinct entities, for the process of growth is a continuum. However, during these periods of development distinct features of growth can be recognized, corresponding with different regulatory mechanisms. Figure 3.1 The infancy, childhood and pubertal (ICP) concept of Karlberg showing the three growth curves. Source: After Karlberg (1989). This can be regarded as a continuation of the foetal growth curve, beginning at conception. The infantile curve is rapid but decelerating, with a mean of 25 cm of growth in the first 12 months of life, and 12.5 cm during the second year. The major regulating influences on growth in infancy are nutritional and thyroidal status. Intrauterine growth restriction (IUGR) results in impairment of the infantile growth curve. The childhood growth pattern starts from approximately six months of age and predominates from the age of three years. During childhood, nutrition becomes less important and hormonal influences, particularly the effects of the growth hormone (GH)–insulin‐like growth factor (IGF) axis and thyroid hormones – become the principal regulating mechanisms for linear growth. Impairment of the childhood growth curve will result from GH or thyroxine (T4) deficiency. During puberty, the pattern of human growth changes dramatically, but differs in important ways between females and males, as described by Tanner (Tanner 1986 – Figure 3.2). Figure 3.2 (a) Height velocity and (b) typical individual height curves in girls and boys throughout childhood and adolescence. Source: (a) from Tanner (1986). In females, the adolescent growth spurt starts approximately two years earlier than in males. Its onset coincides with breast development, the first sign of puberty. Peak height velocity (PHV) occurs on average at approximately 12 years of age and menarche (the onset of menstruation) follows PHV by a variable interval, being close to PHV in early developers and more distant in late developers. Consequently, menarche occurs when height velocity is falling and is followed by approximately two years of gradually diminishing growth rate. A further difference between females and males is the amplitude of PHV which, in females reaches, on average, approximately 8 cm per year compared with 10 cm per year in males. In males, the adolescent growth spurt begins when puberty is already well established and coincides with a testicular volume of 10–12 ml. PHV is reached at an average age of approximately 14 years (15 ml testicular volume). The average difference in adult height between males and females is 12.5–14 cm, (roughly 5 in.) according to UK standards, being accounted for by two additional years of prepubertal growth, a greater amplitude of the adolescent growth spurt, and greater prepubertal height in males. Absence of the pubertal acceleration, with prolongation of the childhood curve, will be seen with delayed puberty. Lack of a pubertal growth spurt despite normal pubertal development is seen in isolated GH deficiency, indicating that both sex steroids and GH are required for a peak to occur. GH is the major endocrine regulator of postnatal linear growth. GH is a single‐chain polypeptide consisting of 191 amino acids, which circulates in the blood bound to growth hormone binding protein (GHBP) that results from cleavage and shedding of the extracellular part of the GH receptor. The predominant form (75%) of GH exists as a 22 kDa protein with 5–10% of pituitary GH release represented by a smaller 20 kDa form that lacks amino acids in positions 32–46. GH is secreted by somatotroph cells of the anterior pituitary gland under the dual regulation of two hypothalamic peptides: growth hormone‐releasing hormone (GHRH), which is stimulatory, and somatostatin, which is inhibitory to GH release. These two peptides are, in turn, influenced by central neurotransmitters. Ghrelin, secreted from the gut, is a third GH secretagogue. GH is secreted in an episodic or pulsatile manner, reflecting the interaction of GHRH and somatostatin. Secretion of GH, mostly via its hypothalamic control, is influenced by a wide variety of environmental, genetic, and physiological factors including nutrition, sleep, exercise, and stress. Figure 3.3 shows the GH‐IGF axis. The insulin‐like growth factors (IGF‐I, IGF‐II) are related peptides, so named because of their close structural relationship with proinsulin and their weak insulin‐like metabolic effects. GH is believed to have little direct effect on linear growth. IGF‐1 is directly responsible for linear growth, particularly during the childhood and pubertal phases. Figure 3.3 The growth hormone–insulin‐like growth factor axis. GH = growth hormone, GHBP = growth hormone binding protein, IGF = insulin‐like growth factor, IGFBP = IGF binding protein. IGF‐1 is primarily secreted by the liver into the circulation and exerts an endocrine effect on target tissues, including bone. IGF‐1 is also synthesized by bone, the cells exerting an influence on adjacent cells – the paracrine effect – as well as on the cells which secrete it – the autocrine effect. The relative contribution of endocrine versus paracrine IGF‐1 on bone remains unclear (Crane and Cao 2014). IGF‐I is a single‐chain polypeptide of 70 amino acids which is encoded from a complex gene on chromosome 12. IGF‐I synthesis occurs after binding of GH to its receptor in the liver and other target organs. The concentration of IGF‐I in the circulation is closely related to physiological secretion of GH, although this relationship may be disturbed in pathological states. IGF‐I interacts specifically with soluble proteins called insulin‐like growth factor‐binding proteins (IGFBPs). The principal carrier protein for IGF‐I is IGFBP‐3, the systemic levels of which depend on GH status. When IGFBP‐3 has an IGF molecule bound to it, it can then associate with another GH‐dependent glycoprotein, ‘acid‐labile subunit’ (ALS), to form a ternary complex. Unlike insulin, most IGF‐I circulates in an inactive or bound form. The IGFBPs extend the half‐life of the IGF peptides, transport them to target cells and modulate their interaction with their respective receptors. IGF‐I binds to the type 1 IGF receptor in target tissues, such as the growth plate at the ends of long bones. Estradiol is the key hormone influencing the GH‐IGF axis at puberty in both sexes (Figure 3.4). The enzyme aromatase is responsible for estradiol synthesis from testosterone in the Leydig cell of the testis; and from androstenedione in the granulosa cell of the ovary. Figure 3.4 Schematic representation of effect of estradiol on growth. Estradiol is aromatized from testosterone in boys and androstenedione in girls. Serum estradiol levels of 15–30 pmol/L in girls and boys are associated with enhanced growth velocity (Albin et al. 2012; Ankarberg‐Lindgren and Norjavaara 2008) via an effect on the growth hormone axis. Larger amounts of estradiol result in growth deceleration and epiphyseal fusion via an effect on the growth plate. The effect of serum estradiol on growth is paradoxical and relates to the level of secretion. Small to moderate levels of serum estradiol accelerate growth by stimulating the hypothalamus to enhance GHRH secretion, resulting in an increase in the amplitude of GH secretion, especially at night (Rose et al. 1991), and increased GH secretion raises IGF‐1 levels. Larger amounts of estradiol inhibit growth through action on the growth plate, accelerating epiphyseal fusion. Thus, estradiol is indirectly responsible for the pubertal growth spurt and directly responsible for the cessation of linear growth in both sexes. See Appendix 1 for Growth Charts. Accurate measurement is essential for growth assessment and an important clinical skill. Errors may be the result of unreliable equipment and faulty measurement technique. To optimize the accuracy of measurement, a single trained measurer – or, at the most two – should measure children in a single clinic. The following techniques enable reliable data to be collected from the most commonly used measurements. Ideally, babies and infants up to the age of two years should be weighed nude, using specialized digital baby/toddler scales. Nappies should always be removed. If the infant is especially fractious and will not lie or sit still enough for a reading to be taken it is acceptable to weigh the parent and child together and then the parent separately and calculate the child’s weight by subtraction. For children over two years, weight should be taken with the subject wearing the minimum of clothing on standing or sitting scales. Where possible, automated equipment should be used and follow national guidelines Measurements should be given in metric units. The cost of equipment for measuring height varies considerably. The stadiometer is recommended for use with children from the age of two years and should be calibrated daily using a metal rod of known length. The subject stands with heels (without shoes and socks), buttocks and shoulder blades against the backplate and the measurer ensures that the imaginary line from the centre of the external auditory meatus to the lower border of the eye socket (the Frankfurt plane) is horizontal. The measurer then applies upward pressure on the mastoid processes and the reading is taken at maximum extension without the heels losing contact with the baseboard. Neonates and toddlers are notoriously difficult to measure accurately. The supine table and neonatometer, both consisting of a flat surface with a fixed headboard and moving baseplate, are two devices developed to reduce error in measuring babies and children too young to stand up (Figure 3.5). Two people are necessary to obtain a reliable measurement. The assistant holds the child’s head in firm contact with the headboard, so that the Frankfurt plane is vertical; with neonates, it is also advisable to use the forefingers to pin the shoulders down. At the same time, the legs of the (by now almost always crying) child are straightened and when the measurer is satisfied that the head is still in contact with the headboard, the measurement can be taken. Figure 3.5 Measurement of supine length. Technically more difficult to measure than standing height, sitting height is required to derive subischial leg length and thus assess body proportion. A sitting height table is required and the subject sits on the table with the back of the knees resting on the edge, with the feet supported on a variable height step so that the upper surface of the thighs is horizontal. The subject is then asked to sit up straight. The headboard is placed on the subject’s head, upward pressure is then applied to the mastoid process and the measurement is taken. Upper arm circumference is not used in routine clinical practice in the developed world but is related to body mass index (Brito et al. 2016) and hence a valuable tool in screening for malnutrition when measurement of height and weight is impractical. The site for measurement is mid‐way between the acromion and the olecranon process. Head circumference is an important measurement of normal growth and should be routine in children under the age of two years. The tape is slipped over the head and passed around the occipital prominence to measure maximal head circumference. Accurate positioning of the tape is vital to ensure reliability and the average of three measurements should be taken. There will always be a degree of intra‐observer error, however competent the operator. With good technique, an error of 0.2–0.3 cm for height measurement by a single observer is achievable (Voss et al. 1990). Expression of age as a decimal makes calculations, particularly height velocity, much simpler. The Table of Decimals of Year, which normally accompanies clinical growth charts, shows what decimal fraction of a year has elapsed by each day. For example, on 2 July 2009 the year has passed 0.501 of the way to 2010 and that date can be expressed as 2009.501. To calculate the decimal age of a child seen at the clinic on 9 May 2018 and born on 26 August 2008, the decimal birthday (108.649) is subtracted from the decimal clinic appointment (118.351) to give a decimal age of 9.702 years. Height velocity should be calculated using measurement intervals of 6–12 months (minimum/maximum 4/14 months), dividing the difference in height (cm) by the difference in interval (years). For example, a child born on 28 March 2002, measuring 132.6 cm on 3 February 2011 and 138.2 cm on 4 January 2012 will have decimal ages at the times of 8.854 and 9.772 so that the height velocity is: An idea of height velocity can also be obtained by examining the growth curve constructed from a series of accurate measurements. A height curve which crosses the centile bands downwards or upwards indicates a change in height velocity from the norm which requires an explanation. Parents should be measured whenever possible. Reported parental heights are notoriously inaccurate, especially in fathers (Cizmecioglu et al. 2005). The mean height difference in men and women from the United Kingdom is 13 cm (Tanner et al. 1970). When a boy is seen in the clinic, the father’s height is plotted directly on the right‐hand side of a growth chart for boys (see Figure 3.6). The mother’s height is then plotted after adding 13 cm to her height. The mid‐parental height (MPH) centile is the mid‐point between the father’s height and the mother’s corrected height. The target range is calculated as the MPH ± two standard deviations = 8.5 cm (6.5–10 cm depending on population data) and represents the 95% confidence limits. Figure 3.6 Growth chart of a boy with constitutional delay in growth and adolescence associated with mild asthma. The father measures 174 cm and his height is plotted directly on the growth chart (F). The mother measures 151 cm and 13 cm are added to her height to correct for the sex difference = 164 cm and this corrected height (M) is plotted. The mid‐parental height (MPH) is 174 + 164/2 = 169 cm with target range ±8.5 cm = 160.5–177.5. Mean (2SD) height prediction (HP) according to the Tanner Whitehouse RUS TW2 system is 162.5 (5) cm. When a girl is seen in the clinic, the mother’s height is plotted directly, the father’s height is plotted after subtracting 13 cm. The MPH and target range are then calculated as for boys. With normal parents and healthy children, the children’s final heights will be normally distributed around the MPH, with only a 5% probability of falling outside the target range. Staging of pubertal development using the criteria of Tanner and the Prader orchidometer in boys is important in the clinical assessment of all patients, irrespective of age. Details of the criteria for pubertal staging are given in Chapter 5. Height for chronological age and bone age can be expressed as standard deviation scores (SDSs) according to the following formula: Height velocity SDS can be calculated according to the following formula: The SDS for height and height velocity at different ages can be obtained by consulting the original publications from which local growth standards have been derived or by downloading software such as Professor Tim Cole’s LMS system, based on UK 1990 data which is available as a free download: http://www.healthforallchildren.com/shop‐base/shop/software/lmsgrowth. Using SDS to express height and height velocity values enables data on groups of children of both sexes and different ages to be pooled and compared statistically. It is also more informative to express extremely short or tall stature as SDS rather than < or ≪ 0.4th centile, > or ≫ 99.6th centile, etc. When SDS values are calculated for patients in puberty, adjustments must be made for the pubertal stage of the child and the age at which PHV occurred. The relationship between weight and height of an individual can be expressed as body mass index (BMI) which is calculated as body weight (in kilogrammes)/height2\(metres). This method has been criticized for assessment of obesity in children because the average values for BMI vary considerably with age. Normal standards for BMI are available for British children using UK 1990 data (Freeman et al. 1995) or since 2009 the UK‐World Health Organization data (see Chapter 11 and Appendix 1 for more information) Several methods have been developed to assess the skeletal maturity or ‘bone age’ of growing children using an X‐ray of the left wrist. The two most commonly used methods are the ‘atlas method’ (e.g. Greulich and Pyle) and the ‘bone‐specific scoring system’ (e.g. Tanner–Whitehouse). Figure 3.6 shows how bone age is represented on the growth chart as an open circle using height at the time of measurement and bone age as coordinates, and linking bone and chronological age with a broken line. An atlas of standard or typical X‐rays of the left hand and wrist of normal girls and boys at specific ages throughout childhood and adolescence is used. The overall standard that most closely resembles the film in question is chosen and this becomes the bone age. Critics of this method claim that a single radiograph may yield bone ages that are several years apart when assessed by different observers. The standards, which are derived from North American children, are also relatively advanced (six to nine months) compared with European children. However, this method is the most widely used throughout the world, with relatively little specialized training being required. In this method, criteria have been established for set stages in skeletal maturation. The most commonly used system is the TW2 method, which also incorporates a methodology for predicting adult height (see Figure 3.6). The system is a bone‐by‐bone, stage‐by‐stage method and the assessor assigns a score to each bone according to written criteria. The composite score is translated into the bone age. This system is generally considered by connoisseurs in the field as being superior to the atlas method as subjectivity is almost eliminated. However, specific training of the assessor is required and use of this method outside the United Kingdom is relatively limited. In 2001, this system of bone age assessment was updated to TW3 to reflect the secular trend towards more rapid physical maturation seen in many countries. In recent years automated systems for bone age assessment have been introduced in clinical practice, with evidence to show comparable accuracy with single‐observer assessment and the advantage of saving labour (Lee et al. 2017). Since 2012, bone age in Glasgow has been calculated using an automated system (http://www.bonexpert.com). Growths charts are compiled using cross‐sectional and/or longitudinal data. Cross‐sectional charts are based on single measurements of large numbers of individuals, covering the whole age range. Longitudinal charts are based on regular, serial measurements of a smaller number of individuals. Three main types of growth charts are currently available in the United Kingdom. The UK 90 charts published by the Child Growth Foundation in 1990 are cross‐sectional and were compiled using contemporary UK data. They incorporate a novel nine‐centile format with each centile ‘band’ being 2/3 SDS apart. The resultant growth curves are similar to the conventional 3rd–97th centiles, with additional curves −2.67 SD (0.4th centile) and + 2.67 SD (99.6th centile) about the mean to give more useful cut‐offs for stature screening. The Buckler‐Tanner charts (1995) are a revised version of the former Tanner‐Whitehouse charts and incorporate both cross‐sectional (in the childhood curve) and longitudinal (in the pubertal curve) data, making them particularly useful for the interpretation of serial measurements. In 2010, UK–WHO growth charts for the age range birth to four years were introduced. These charts are based on the World Health Organization (WHO) international child growth standards which describe the optimal growth of healthy breast‐fed children from six countries. In 2012, the Royal College of Paediatrics and Child Health (RCPCH) among other organizations recommended use of the 2–18 year WHO growth charts (www.rcpch.ac.uk/resources/uk‐world‐health‐organisation‐growth‐charts‐2‐18‐years). In the USA, many of the major centres use growth charts incorporated into electronic medical record systems. The National Center for Health Statistics (NCHS) Centers for Disease Control and Prevention (CDC) growth charts and the WHO growth charts are widely used. NCHS recommends the WHO growth charts for children aged 0–2 years and the NCHS (CDC) charts for children aged 2 years and older (https://www.cdc.gov/growthchart /). In Canada, the Canadian Pediatric Endocrine Group (CPEG) officially endorsed the 2014 revision to the WHO Growth Charts for Canada, with a preference for the use of the Set 2 charts (https://www.cpeg‐gcep.net/content/who‐growth‐charts‐canada). For clinical use we find that the split format of the WHO charts is not ideal for monitoring, and demonstrating to families, the growth status and height trajectory of patients with problems such as constitutional delay, growth hormone deficiency and congenital adrenal hyperplasia. The UK editors prefer the Child Growth Foundation (CGF) and Buckler‐Tanner charts for children aged two years or more with endocrine and growth problems while the North American editors often use the Tanner and Davies longitudinal growth charts (Tanner and Davies 1985), especially for children with short stature and delayed onset of puberty. There is agreement that the WHO charts are indicated for children aged 0–2 years. Appendix 1 therefore reproduces the WHO 0–4 year and CDC 2–20 year height and weight charts; the Tanner and Davies 2–19 yearheight velocity charts; and the UK‐WHO body mass index (BMI) 2–20 year charts for girls and boys. In addition, Appendix 2 shows disease‐specific growth charts for Turner, Down and Noonan syndromes, and achondroplasia. Much of this is within the scope of all healthcare professionals who deal with children, including family practitioners, school nurses, and health visitors. With a combination of meticulous auxology, thorough history, and focused examination, a sensible diagnosis and management plan can be achieved. The following should be carried out before the child/adolescent and family enter the consulting room: The most likely diagnosis and differential diagnosis should be formulated before any investigations are contemplated. This is unnecessary in most short children, for example, those with normal familial short stature and/or constitutional delay in growth and adolescence. Clinical features suggesting that investigation of short stature is indicated are given in Table 3.1. Investigation should always be seriously considered in a child whose height centile falls below the parental target range centiles. Table 3.1 Clinical features suggesting that investigations for short stature are indicated. Once the decision has been made to perform laboratory investigations on the child with short stature, the doctor must proceed as a general paediatrician and not as a paediatric endocrinologist. If the approach is too specialized, disorders such as anaemia, malabsorption, renal disease, Crohn disease, or even Turner syndrome can easily be missed. Baseline investigations for short stature are shown in Table 3.2. Note that no investigations of GH status other than IGF‐1 are included at this stage. Table 3.2 Baseline investigations for short stature. The decision to investigate GH secretion is made only after the above investigations have been performed and documented to be normal. At the second and third consultations additional auxological information, particularly on height velocity, will be available. Further investigation of the child can now be considered. Indications and procedures for investigation of possible GH insufficiency are covered in section on the diagnosis of GH deficiency, which discusses this disorder in detail. Short stature may result from a combination of factors (composite short stature) rather than a single cause. For example, a child might be short due in part to IUGR but may also have short parents, poorly controlled asthma, and psychosocial difficulties. A suggested classification of the major aetiological categories is shown in Table 3.3. This classification separates idiopathic short stature (ISS) from normal familial short stature. There is some overlap between categories; some dysmorphic children are small‐for‐gestational age (e.g. foetal alcohol syndrome), while skeletal dysplasia is a component of other syndromes (e.g. Turner syndrome). Table 3.3 Classification of short stature. This is the most common cause for referral of a child with short stature. Essentially, the child is perfectly healthy but has inherited short stature genes from one or both parents or, occasionally, a more distant relative. There is no endocrine abnormality and the bone age is usually not delayed, unless there is also a component of growth delay. NB: Do not assume that familial short stature is always normal. If one of the parents is markedly short, a dominantly inherited growth disorder, such as a skeletal dysplasia, neurofibromatosis, or GH deficiency, should be considered. The diagnosis of constitutional growth delay (see Figure 3.6) is suggested when the child is healthy but looks young for his/her age, has evidence of late maturation in terms of bone age and pubertal delay, and often the history that one parent or second degree relative (e.g. an uncle) was short during childhood with subsequent delay in puberty. A history of atopic asthma, often mild, is common. The condition is seen more often in boys and typically there is a component of genetic short stature since it is the shorter children who tend to show slow maturation. Frequently, the slow maturation starts in early childhood and the delay of physical development accumulates. Consequently, an 11‐year‐old boy could well have the physical maturity, bone age, height, and appearance of an 8‐year‐old. The short stature in this situation is then compounded by the delay in pubertal development. Typically, final adult height is within but in the lower half of the parental target range (see Figure 3.6). Smallness‐for‐gestational age (SGA) is linked with, but distinct from, intra‐uterine growth restriction (IUGR) (see Figure 3.7). SGA is defined according to weight and varies with the paediatric discipline involved. Neonatologists define SGA as birth weight below the 9th or 10th centile for gestational age, recognizing that this population is at risk of postnatal hypoglycaemia, and should be targeted for early feeding and capillary glucose monitoring. Endocrinologists define SGA in auxological terms – birth weight below the 2nd or 3rd centile (i.e. more than 2 SD below the mean). Using the latter definition of SGA will include many small normal babies. Short stature at birth can only be reliably identified if birth length is measured accurately, and is only roughly correlated with birth weight (Sardar et al. 2015). Figure 3.7 Intrauterine growth chart showing three patterns of intrauterine growth. The solid line shows impaired foetal growth with birth length/weight below the – 2 standard deviation (SD) curve, i.e. both small for gestational age (SGA) and intrauterine growth restriction (IUGR). The broken dashed line shows normal growth below but parallel to the – 2 SD curve, i.e. SGA but no IUGR. The dotted line represents impaired foetal growth but birth weight/length is appropriate‐for‐gestational age (AGA), i.e. IUGR but no SGA. IUGR means intrauterine growth failure. Affected infants are usually but not always light/short at birth (see Figure 3.7). The diagnosis of IUGR is secure if serial antenatal ultrasound has demonstrated growth failure; and in defined syndromes such as Russell‐Silver; likely in the context of severe SGA; and can be inferred in SGA or non‐SGA infants with birth weight < 25th centile and a combination of postnatal feeding difficulties and impaired infantile growth curve. If weight, length, and head circumference are proportionate at birth, the SGA is termed symmetrical and reflects either a small/normal baby or IUGR related to early growth failure. Asymmetrical SGA, when weight is reduced but with preservation of head and linear growth, occurs because of late deprivation of nutrients usually related to placental insufficiency and is the foetal equivalent of failure to thrive. Babies with asymmetrical SGA are more likely to show postnatal catch‐up growth. The overall catch‐up rate within two years of birth in SGA subjects is 87% (Albertsson‐Wikland and Karlberg 1994). SGA also can be classified according to the cause: Dysmorphic syndromes can be defined as conditions arising from a specific event – chromosomal, genetic, environmental – during foetal life and resulting in a variable constellation of features (Jones 1997). These include: Diagnosis is important not only to provide a cause for the short stature but also to identify associated problems (e.g. ovarian failure in Turner syndrome) and giving the patient and family a prognosis. The paediatrician seeing children with short stature needs to become experienced in recognizing clinical patterns suggestive of a dysmorphic process, and being aware of the heterogeneity of each syndrome. Professor James Tanner taught that identification of dysmorphic features is easier with the patient standing opposite the seated doctor. Every patient in Professor Tanner’s clinic was photographed, which also accentuated unusual features. The involvement of a clinical genetic colleague is invaluable, particularly in assessing the many patients with dysmorphism who do not fit into an obvious diagnostic category. A detailed account of all the relatively common recognizable dysmorphic syndromes is beyond the scope of this chapter; however, a selection of six syndromes which may present undiagnosed to the growth clinic are briefly described here. Birth weight is usually <2nd centile and almost invariably <9th centile, feeding difficulties are the rule, and nocturnal sweating (suggestive of hypoglycaemia) is common. On examination, affected individuals are short and thin, some showing asymmetry with hand, foot, arm, or leg shorter on one side. Head is relatively large, often with expanded vault, and facies are characteristically triangular with down‐sloping eyes. Intelligence is normal. The condition results from maternal and/or paternal iso‐ and heterodisomy causing abnormal methylation in imprinted genes. Causes include, but are not confined to, hypomethylation at 11p15.5 and to maternal disomy of chromosome 7 (Dias et al. 2012). Hypomethylation at 11p15.5 has been shown to affect postnatal mRNA expression of IGF‐2. Birth weight is invariably reduced, and there is postnatal short stature associated with poor concentration, behaviour and learning difficulties. Facies are variable but may show smooth philtrum, short palpebral fissures, and malar hypoplasia. Head circumference is reduced. A history of maternal alcohol ingestion is crucial to making the diagnosis but is not always evident at initial presentation. Hence, SGA/IUGR children with developmental delay and poor concentration should be followed up so that foetal alcohol spectrum disorder (FASD) can be recognized. Turner syndrome (see Table 3.4 and Figure 3.8) occurs in about 1 in 2500 liveborn girls. There is loss or abnormality of the second X chromosome in at least one major cell line in a phenotypic female. The principal features are outlined in Table 3.4 and include dysmorphic traits (see Figure 3.8), short stature (an almost constant sign) and ovarian dysgenesis (in about 90%). Associated features include congenital heart disease (15%), middle ear disease which can be very troublesome (65%), hypertension and aortic root dilatation, renal anomalies (common but rarely problematic), a predisposition to autoimmunity, specific learning difficulties (particularly with mathematics), and a degree of social vulnerability. Table 3.4 Principal features of Turner syndrome. Figure 3.8 Three‐year‐old child with Turner syndrome showing ‘shield’ chest and relatively short legs. Partial inactivation of the second X chromosome in all the body’s cells from early foetal life partly explains the remarkably mild phenotype of Turner syndrome. However 30% of the short arm genes of the second X chromosome are not inactivated, and these include the Short Stature Homeobox (SHOX) gene. Loss of this and other genes results in skeletal dysplasia with short stature. Loss of genes controlling lymphogenesis, ovarian function, and skin naevi result in lymphoedema, accelerated atresia of oocytes, and increased naevi (particularly on the face), respectively. The second chromosome may be completely lost (45,X), undergo duplication of the long arm (q) with concomitant loss of the short arm (p) to form an isochromosome (isoXq), undergo ring formation (rX), or deletion in the short or long arm (Xp− or Xq−). Complete 45,X monosomy accounts for 40–60% of the karyotypes on peripheral blood lymphocytes, while most of the remaining karyotypes show a mosaic pattern; for example, 45,X/46,XX, 45,X/46,X,iXq, 45,X/46,XY, 45,X/46,X,rX. There is little specific phenotype/genotype correlation for most karyotypes but phenotype is milder in the 45,X/46,XX and 45,X/47,XXX variants. All three components of Karlberg’s ICP model are impaired in Turner syndrome, with a degree of IUGR (usually mild) in almost all cases; subnormal growth during infancy and childhood with 15 cm loss in height compared with normal girls between the ages of 3 and 12 years; and impaired pubertal growth spurt even with oestrogen therapy, related to the underlying skeletal dysplasia. Adult height in European populations ranges from 142 to 147 cm (Ranke 1998). There is a positive correlation between adult height and parental height. Because of the subtlety and variable incidence of many of these features, Turner syndrome is easily missed. The diagnostic key is short, or borderline short, stature in a girl whose height is inappropriate for the parental target range centiles (Ouarezki et al. 2017). Karyotype analysis is indicated in this situation and/or in the context of dysmorphic features, middle ear disease, and cardiac anomalies. Follow‐up is required to pre‐empt associated problems such as autoimmune thyroiditis, middle‐ear disease, learning difficulties, and hypertension and is best provided in designated Turner syndrome clinics, with good hand‐over arrangements for specialist adult care. Noonan syndrome (see Table 3.5, Figure 3.9 and Appendix 2) refers to a heterogeneous group of conditions resulting from various gene mutations, of which four have been identified – PTPN11 (encoding SHP‐2), SOS1, KRAS, and RAF‐1. Sporadic mutations are commonest, affected individuals then transmit the condition in autosomal dominant fashion (see Figure 3.9). Prevalence is estimated at between one in 1000–2500 live births. The principal features of Noonan syndrome are shown in Table 3.5. Growth is usually affected with height velocity being subnormal and puberty is typically delayed and associated with a blunted adolescent growth spurt. Adult heights are approximately 162 cm in males and 152 cm in females (Ranke et al. 1998). Table 3.5 Principal features of Noonan syndrome. Figure 3.9 Father and twin boys with Noonan syndrome. This condition arises from a spontaneous deletion affecting 7q11.23 and may present with short stature in a child with intellectual disability but with aptitude for music, increased fearfulness, engaging behaviour described as ‘cocktail party manner’, hyperacusis, and characteristic facies – button nose, full lips, and ‘elfin’ appearance. Associated problems include infantile hypercalcaemia, supravalvular aortic stenosis, pulmonary artery stenosis, hypertension, and scoliosis. This condition results from a mutation in the FGDY1 gene located on Xp11.21. The phenotype – hypertelorism, interdigital webbing, short broad hands, and shawl scrotum in males – is milder in affected females and more pronounced in their sons. Intelligence is normal and the degree of short stature usually insufficient to warrant intervention. This is a varied and complex group of disorders requiring input from colleagues in other disciplines including genetics, radiology, and orthopaedics, as well as endocrinology. Patients may present with severe short stature and obvious disproportion (abnormal upper:lower segment ratio, sitting height, and arm span relative to height), in which case the diagnosis of skeletal dysplasia is obvious. Less severe cases may present with short stature of uncertain cause requiring a diagnosis, and shrewd assessment may be needed to detect mild disproportion. The limbs will be short in disorders principally affecting the long bones, for example, achondroplasia, hypochondroplasia, and metaphyseal dysplasia; the back will be short in disorders affecting the spine as well as the long bones, for example, spondylo‐epiphyseal‐dysplasia (SED). Table 3.6 shows the key features of three important disorders – achondroplasia, hypochondroplasia, and spondylo‐epiphyseal‐dysplasia. The following points should be borne in mind: Table 3.6 Key features of achondroplasia, hypochondroplasia, and spondylo‐epiphyseal‐ dysplasia. Chronic illness is a potent cause of impaired linear growth in childhood and adolescence, and pubertal delay (Kao et al. 2018). The child may already be diagnosed, and referred to evaluate the contribution of various components including the disease (e.g. chronic arthritis) and its treatment (e.g. systemic steroids) to the poor growth. By contrast, chronic disease may be an unsuspected cause of short stature. Gastrointestinal disorders, such as coeliac disease and inflammatory bowel disease, can present in this way, while chronic renal disease is notoriously silent. The degree of growth failure varies considerably from a relatively mild effect, usually with constitutional delay, in mild to moderate asthma to potentially severe short stature in Crohn disease and juvenile chronic arthritis. The three principal mechanisms for poor growth and short stature in chronic disease are: Other factors include delayed maturation and puberty; hypoxia (cyanotic congenital heart disease); metabolic disturbance (glycogen storage disease, renal failure); and partial GH resistance (Crohn disease, renal failure) There is an established relationship between socioeconomic status and physical growth. However, it can be difficult to weigh up the contribution of the social environment to short stature and poor growth in the individual. This is because factors such as SGA, poor diet, respiratory illness related to parental smoking, and parental short stature may be contributory and of variable relation to the poor social circumstances. Adverse family and social factors can delay a child’s physical and emotional development. David Skuse and colleagues described a striking variant of psychosocial growth failure featuring abnormal behaviour with hyperphagia and polydipsia. Investigation shows GH insufficiency which is resistant to exogenous GH therapy but is reversible on moving the child to a favourable environment (Skuse et al. 1996). Psychosocial deprivation should always be considered when a child is referred with short stature, and the clinician must take a thorough family and social history. Careful follow‐ up of suspected cases, with sequential height and weights, is crucial not only in confirming the diagnosis, but also in documenting the child’s needs in terms of future care and placement. These are controversial! GH insufficiency can be defined in terms of a peak GH level which is below an agreed arbitrary cut‐off level. Assays vary between centres while the intra‐patient variability of peak GH secretion is high. Severe GH insufficiency can be defined as a maximum stimulated GH concentration of <3 μg/L), partial GH insufficiency as 3‐7μg/L. It is important to recognize that low GH levels can be encountered in virtually any short child and that low levels are not indicative of permanent impairment of the hypothalamo–pituitary (H‐P) axis. It follows that GH status should be measured judiciously and carefully interpreted in the clinical context. A significant proportion of peripubertal children with short stature, slow height velocity, and low stimulated GH levels will show normal GH status on retesting once final height is attained. In retrospect, many such children would have been displaying constitutional delay with hypothalamo–pituitary ‘dormancy’. The term GH deficiency (as opposed to GH insufficiency) can be used to refer to permanent impairment of the GHRH‐GH axis, due to either congenital or acquired disease. According to this nomenclature, therefore, GH deficiency is a subset of GH insufficiency. We recommend using the term ‘GH deficiency’ if actual impairment of GH secretion is either certain or very likely, and the term ‘GH insufficiency’ when GH levels are low but definite H‐P axis abnormality has not been demonstrated. While classic and severe growth hormone deficiency is clinically easy to diagnose (see Figure 3.10), milder forms are difficult to separate from normal variant short stature. The features of these two ends of the spectrum are shown in Table 3.7 and the causes of GH deficiency are shown in Table 3.8. Figure 3.10 Five children aged two to seven years from two consanguineous families (two brothers married two first cousins who were sisters) with severe isolated GH deficiency. One parent from each family is shown in the picture. Note the excess subcutaneous fat, immature appearance and small genitalia in the affected boys. Source: Donaldson et al. 1980, reproduced with kind permission of British Journal of Medical Genetics. Table 3.7 Clinical features of severe and mild GH deficiency. Abbreviations: ACTH, adrenocorticotrophic hormone; FSH, follicle‐stimulating hormone; GH, growth hormone; LH, luteinizing hormone; TSH, thyroid‐stimulating hormone. Table 3.8 Principal causes of GH insufficiency. In >80% of cases, GH insufficiency is ‘isolated’, i.e. not associated with other anterior pituitary hormone deficiencies and may (see above) turn out to be a transient insufficiency rather than a permanent deficiency, falling into the ‘mild’ rather than ‘severe’ category described above. It is now known that the basic defect in most true isolated GH deficiency cases is in the synthesis or release of the hypothalamic peptide GHRH, but the precise pathogenesis is unknown. However, these patients respond to administration of exogenous GHRH by secreting GH, indicating that the somatotroph cells are functional and that the primary defect is in the hypothalamus. Magnetic resonance imaging (MRI) imaging and genetic studies are normal. True isolated GH deficiency related to genetic and structural MRI abnormalities presents as: Figure 3.11 MRI scan showing ectopic posterior pituitary gland (1) and small anterior pituitary (2) in a 15‐year‐old girl with GH deficiency and partial gonadotrophin deficiency. Of these three features, EPP has the lowest inter‐observer variability and is the most strongly predictive of GH deficiency. Multiple anterior pituitary hormone deficiencies may present in the newborn period or early infancy with: Hypothyroidism of pituitary origin is likely to be present, becoming manifest later, but usually without intellectual impairment. Low serum IGF‐I and IGFBP‐3 levels may be suggestive of GH deficiency. Subsequent imaging may show small anterior pituitary/interrupted stalk/EPP. The treatment of congenital hypopituitarism is a neonatal emergency. It is crucial to give hydrocortisone to prevent persisting hypoglycaemia. If this is not effective, GH therapy must be added. T4 replacement will also be indicated. GH reserve should not be tested, as GH stimulation tests can induce serious and life‐threatening hypoglycaemia. Gene mutations affecting the pituitary transcription factors Pit‐1 (causing GH, thyroid‐stimulating hormone [TSH], and prolactin deficiency) and Prop‐1 (deficiency of GH, ACTH, TSH, LH, follicle‐stimulating hormone [FSH]) are now well documented in multiple pituitary hormone deficiencies. Genetic defects are rarely found in the structural central nervous system (CNS) defects. Congenital CNS defects occurring in the mid‐line may be associated with GH deficiency, usually with multiple pituitary hormone deficiencies. However, these lesions cause considerable endocrine heterogeneity. The most frequent is the syndrome of septo‐optic dysplasia consisting of two or three components of the triad: GH deficiency may be isolated, and evolve during childhood. ACTH and TSH deficiencies are common, vasopressin deficiency affects 20%. Interestingly, LH and FSH secretion is often preserved (and there may even be sexual precocity) but some patients have partial or severe deficiency. Presentation is usually in early infancy with either visual abnormality (roving nystagmus and failure of fixation), or hypopituitarism (neonatal hypoglycaemia, jaundice, poor feeding). Learning disability is a variable associated feature, some patients being of completely normal intelligence, others severely affected. Defects in the HESX‐1 gene have been described in a few cases, mainly familial in nature. Other congenital defects which can be associated with GH deficiency are agenesis of the corpus callosum, holoprosencephaly, and arachnoid cysts. These conditions can be diagnosed by MRI scan; the risks of general anaesthetic in the hypopituitary infant must be carefully considered. Although rare, this is the most common tumour in the hypothalamo‐pituitary region in childhood. Although histologically benign, craniopharyngioma is locally invasive, involving adjacent structures, including the hypothalamus, thus affecting the pituitary function, and the optic nerves, and chiasm. The three modes of presentation are: The management of craniopharyngioma is complex and controversial. The devastating endocrine and psychoneurological morbidity of radical surgery with the removal of hypothalamic tissue is well recognized (De Vile et al. 1996). Complete macroscopic removal is now attempted only if damage to adjacent structures can be avoided. Otherwise the tumour is decompressed and debulked as safely as possible, following which radiotherapy is given. This may present with diabetes insipidus alone for many years before the tumour itself and other pituitary deficiencies become manifest. Consequently, so‐called ‘idiopathic’ diabetes insipidus must always be viewed with suspicion and investigated with serial MRI of the brain. Bifocal germinoma with suprasellar and pineal lesions may occur. Pituitary stalk thickening may be the first radiological abnormality. Elevation of serum ± cerebrospinal fluid human chorionic gonadotrophin (hCG) and α‐fetoprotein (AFP) levels are useful tumour markers. Treatment is with chemotherapy and craniospinal radiotherapy. Optic nerve glioma, which occurs more commonly in patients with neurofibromatosis, may also be associated with pituitary deficiency and, paradoxically, precocious puberty. Targeted radiotherapy is indicated if vision is threatened but most cases are managed conservatively, giving endocrine therapy as required. The infiltrative lesion of Langerhans cell histiocytosis typically involves the hypothalamus and causes diabetes insipidus. In a proportion of cases, this will be associated with GH deficiency. This topic is dealt with in Chapter 12. Children who have received CNS irradiation, whether for prophylaxis for leukaemia, for tumours distant from or adjacent to the hypothalamo‐pituitary region or during total body irradiation are at risk for the development of GH deficiency, and should receive endocrine monitoring and follow‐up. As indicated earlier, most children with GH insufficiency do not have ‘severe’ GH deficiency (see Table 3.7). A combination of both auxological and biochemical criteria is required to make this diagnosis. GH therapy should not be prescribed without documentation of biochemical GH insufficiency. GH profile. GH secretion is pulsatile, with peaks occurring approximately every three hours. A pattern of GH secretion, demonstrating physiological peaks and troughs can be obtained by continuous or 20‐minute venous sampling for serum GH levels through an indwelling cannula. This technique, known as GH profiling, is too time‐consuming, labour‐intensive, and expensive to be recommended for routine clinical practice but remains a valuable research tool. Serum markers of GH secretion or action. Serum IGF‐I and IGFBP‐3 reflect the status of GH secretion, provided that the GH receptor is functioning normally. In severe GH deficiency, IGF‐I and IGFBP‐3 levels are low; however, the large overlap between normal children and children with less severe GH deficiency limits the diagnostic value of IGF‐1. Also, low IGF‐I in a short child may reflect a negative caloric balance (e.g. in children treated with psychostimulant medication) rather than GH deficiency. GH stimulation tests. This difficult area has been addressed in the 2016 guidelines by the Paediatric Endocrine Society (Grimberg et al. 2016) and by the British Society for Paediatric Endocrinology and Diabetes (BSPED GH standards document, 2017). The former guidelines caution against ‘reliance on GH provocative test results as the sole diagnostic criterion of GHD’. BSPED guidelines recommend that GH stimulation testing should only be carried out by experienced personnel in centres carrying out a minimum of 10 tests per year. GH stimulation testing is not needed in patients who satisfy these three conditions: (i) auxological criteria for GH deficiency (particularly low height velocity) are met; (ii) MRI scan shows one of the classic triad of defects, or presence of tumour or irradiation affecting the HP‐axis; and (iii) at least one other anterior pituitary hormone deficiency has been demonstrated. Neither is testing recommended in newborns with serum GH < 5 μg/L in the context of hypoglycaemia plus either deficiency of at least one additional pituitary hormone; or one or more classic features on MRI imaging; or both. In other situations, TWO stimulation tests are recommended, five of which are shown in Table 3.9. Table 3.9 Details of some GH stimulation tests. Absolute requirements before all GH stimulation tests are to document normal serum T4 concentration and normal serum cortisol concentration (>100 nmol/L). Note that glucagon and insulin tolerance tests also test adrenal axis while additional cortisol testing is required for the other three methods. Testing is performed after an overnight fast. Abbreviations: GH, growth hormone. Between about nine years and the onset of the pubertal growth spurt, the hypothalamo‐pituitary axis has become relatively ‘dormant’ so that height velocity slows and response to GH stimulation is diminished. Boys and girls with constitutional delay may be mis‐diagnosed as having isolated GH deficiency on direct stimulation testing and are subsequently shown to have normal pituitary function on retesting once pubertal development is complete. This diagnostic trap may be offset by ‘priming’ the axis with sex steroids prior to stimulation testing from nine years until Tanner stage G3 in boys and Tanner stage B2 in girls. The counter‐argument for carrying out sex steroid priming is that children with functional growth hormone insufficiency during the peripubertal years may benefit from GH treatment both in terms of enhancing short‐to‐medium term growth; and improving final height. There is thus a diagnostic argument in favour of priming and a therapeutic argument against priming. Given the expense of growth hormone, and the concerns about subjecting essentially healthy subjects to years of daily injections, we favour sex steroid priming. The two most commonly used tests in the United Kingdom are the glucagon and clonidine tests. The glucagon test also stimulates cortisol secretion, which can be an advantage if multiple pituitary hormone deficiencies are suspected but may cause hypoglycaemia, particularly in young children. The insulin‐tolerance test (ITT) is avoided by many centres in the United Kingdom because of the risks of serious hypoglycaemia, and should never be used for children under five years, nor should it be performed in a non‐specialist environment. However, in the context of an established paediatric endocrine unit, the ITT has been found to be safe and probably provides the best validated stimulus for GH secretion. Whichever test is used, secure intravenous access with a good‐sized cannula in situ, oxygen and suction facilities, and experienced staff must be available. If hypoglycaemia occurs, the symptoms can be quickly and safely relieved with oral or intravenous administration of glucose. This is discussed in Chapter 6. Unless diagnosed early, congenital hypothyroidism leads to severe stunting of growth. The introduction of neonatal screening has eliminated this cause of short stature. By contrast, acquired hypothyroidism caused by autoimmune thyroiditis may present with short stature and growth failure, hence the need to include thyroid function testing in all short children requiring investigation. Hypercortisolaemia impairs linear growth. Consequently, most patients with Cushing’s syndrome will have a deceleration in height gain, in marked contrast to subjects with simple obesity in whom height velocity is enhanced. Exceptions are cases of Cushing’s syndrome where excess adrenal androgens are secreted, which may counteract the growth‐suppressive effect of high cortisol. Cushing’s syndrome is discussed in Chapter 8. GH resistance accounts for a very small number of patients with short stature. In its severe form, it presents as Laron syndrome, a rare severe autosomal recessive disorder caused by a homozygous mutation of the GH receptor. Growth failure is extreme in this condition, with an untreated adult height of 120–130 cm. Milder forms of GH resistance may be a cause of short stature, but this has yet to be established. This is a descriptive rather than a diagnostic category and its definition is controversial. In this chapter, ISS refers to children with height < −2.5 SD in whom the problem is either not attributable to normal familial short stature or constitutional delay, and in which other causes of short stature have been excluded. ISS probably includes a range of conditions including partial GH resistance and subtle skeletal dysplasias which cannot be detected on standard skeletal X‐rays. A limited repertoire of growth‐promoting preparations is available for short stature treatment: growth hormone; the weakly androgenic anabolic steroid Oxandrolone; and the sex steroids. GH is licensed for treatment of GH insufficiency, Turner syndrome, Prader‐Willi syndrome, short SGA children, and short children with renal failure. Oxandrolone can be used for growth promotion in peripubertal boys and in Turner syndrome. Sex steroid therapy is available for stimulation of pubertal growth. Other therapies, such as GHRH and IGF‐I, are not widely used in routine practice. The cornerstone of management is correct diagnosis, reassurance, and a realistic prognosis for final height, the latter being facilitated by adult height prediction in subjects aged more than 10–11 years. Some families with constitutional growth delay can be discharged after the initial consultation. Others find it helpful to receive regular 6‐monthly follow‐up until the pubertal growth spurt is well underway. In roughly half the boys seen with constitutional delay, there is no strong demand for therapy, but Table 3.10 shows some features which guide towards intervention. Table 3.10 Indications for consideration of endocrine therapy in the patient with constitutional delay in growth and adolescence. The aims of treatment depend on the age of the patient: growth acceleration in boys aged 10–13 years; and both growth and pubertal acceleration > 13 years. Oxandrolone is helpful in accelerating growth in boys aged >10 years who are too young for puberty to be induced. Unfortunately, it has not been manufactured in Europe since 2008 but it can be purchased from North America for use in selected cases. Oxandrolone is given in the dose of 1.25 mg orally at night for three to six months in boys aged <12 years and 2.5 mg for boys aged >12 years. Testosterone esters will accelerate growth and pubertal development. This therapy does not affect final height and simply speeds up the pubertal process. Testosterone cypionate (Sustanon) or enanthate (Primoteston Depot) 50–125 mg by intramuscular injection every four weeks for three to six months are available for use. Testosterone enanthate 125 mg by intramuscular injection for three months only was shown in a Scottish study to be effective and to have no adverse effect on final height in comparison with untreated boys (Kelly et al. 2003). There is a good case therefore for giving testosterone in this dose rather than using lower, less effective doses such as monthly injections of 50 mg. Oral testosterone undecanoate 40 mg daily, increasing to 80 mg daily depending on response, is an effective alternative to intramuscular testosterone (Lawaetz et al. 2015). Endogenous puberty can be followed by serial measurement of testicular volumes which are unaffected by exogenous testosterone and reflect gonadotrophin activity. If the testes remain at <5–6 ml a year after testosterone treatment, indicating that endogenous puberty is still in the early stages, a second course of testosterone may be given. In girls with delay of growth and puberty, oral ethinyl‐estradiol 2 μg either daily or on alternate days for 6–12 months will induce some growth acceleration, which may be associated with early breast development. However, careful patient selection and evaluation are required in this situation, some specialists electing to ‘cover’ oestrogen treatment with GH therapy. GH insufficiency can be effectively treated with recombinant GH. Successful treatment is potentially able to normalize height, but this is achieved at a significant economic cost and requires daily subcutaneous injections. The less severe the GH insufficiency, the less the benefit from replacement therapy. Every child considered for GH therapy must be assessed in detail so that treatment is reserved for those who will unequivocally benefit by an increase in final adult height. Early diagnosis and initiation of therapy in GH deficient children must be the goal of all those involved in growth assessment. Before starting treatment, auxological assessment for 6–12 months is desirable unless the time available for further growth is limited, or the degree of GH deficiency and short stature is severe. The following conditions should be met: Table 3.11 Guidelines for GH therapy in children with GH insufficiency. The role of the endocrine nurse specialist is crucial in: Three‐ to four‐monthly, rather than six‐monthly clinic visits are recommended to adequately monitor the response to and compliance with treatment. Measurement of IGF‐1 in children receiving GH is a yardstick against which compliance/adherence can be monitored. IGF‐1 measurement is also advised for safety, decreasing the GH dose if the IGF‐1 level is >2 standard deviations above the age‐related laboratory reference range. Increasing GH dosage at puberty to mimic the physiological increase that occurs during this phase seems logical but is controversial. FDA recommendations are for an increase from 30–50 to 100 μg/kg/day to cover the pubertal years while the Paediatric Endocrine Society guidelines advise against the routine increase in GH dose during this phase of growth. After linear growth is complete, appraisal of GH status should be according to consensus guidelines (Clayton et al. 2005). When permanent deficiency is certain, e.g. with craniopharyngioma, genetically proven GH deficiency and multiple pituitary hormone deficiency, there is no need to stop treatment. If GH deficiency is highly likely, an IGF‐1 level is taken six weeks after stopping treatment, a subnormal value being sufficient to confirm the diagnosis. Otherwise, a formal GH stimulation test is required, using insulin, arginine or glucagon but not clonidine. Many patients with ‘idiopathic GH insufficiency’ will now have a normal response and can be retrospectively diagnosed as having had constitutional delay. Subjects with proven GH deficiency should be transferred to an adult endocrine clinic for follow‐up. Experience has shown that a transition clinic attended by both paediatric and adult endocrinologists improves the quality of transfer, and the chance of compliance with attendance at the adult clinic. Recombinant GH has a remarkably good safety record. The complication of Creutzfeldt‐Jakob disease associated with pituitary‐extracted GH was identified in 1985 and from this time only biosynthetic GH should have been used. Intracranial hypertension, usually seen shortly after the initiation of therapy, resolves with temporary cessation of GH followed by reintroduction at a lower dose. Slipped capital femoral epiphysis and progression of pre‐existing scoliosis should be detected by clinical awareness and monitoring, including regular examination of the spine. Any increase in cancer risk among GH‐treated patients can be largely related to previous treatment for malignancy, and a direct influence of GH is doubtful. A large study of nearly 24 000 GH‐treated patients from eight European countries showed no general increase in cancer risk, but the finding of a slight increase in bone cancer, bladder cancer, and Hodgkin’s lymphoma requires ongoing surveillance (Swerdlow et al. 2017). The availability of recombinant GH, although admittedly at high cost, has led to its use in a number of non‐GH‐deficient disorders, where its pharmacological properties might benefit growth. Prominent among these are Turner and Noonan syndromes, short stature related to SGA, renal disease, and skeletal dysplasias. Each of these categories will be discussed briefly. The use of GH therapy in children with ISS remains controversial and often presents the clinician with a moral dilemma. The efficacy of this treatment has been demonstrated, with several clinical studies showing a positive effect on final height. Indeed, GH therapy was approved in the US for children with this indication in 2003. In the United Kingdom, this indication has not found favour, experience showing that many of the families whose children have short stature and borderline GH levels (which do constitute an indication for treatment) become discouraged by the modest increase in height status after one to two years, and either openly state their wish to discontinue treatment, or demonstrate their lack of enthusiasm by defaulting from the outpatient clinic. While optimizing growth and puberty in Turner syndrome is important, it is essential that other aspects of this complex condition, including cardiovascular, otological, bone, uterine, and psychological health, are not neglected and are well reviewed in the 2017 Cincinnati guidelines (Gravholt et al. 2017). Mildly affected girls (e.g. some patients with 45,X/46,XX mosaicism) may not require growth‐promoting treatment, but short stature is usually sufficiently severe for most families to opt for intervention. GH has been definitively shown to increase final height by approximately 7 cm (Stephure 2005). However, all studies have demonstrated varying responses in individuals, some girls faring much better than others. Because of the skeletal dysplasia component in Turner syndrome, the GH dose required is higher than for classical GH deficiency – 55 μg/kg/day (10 mg/m2/week).GH can be started when the patient’s height falls below the 3rd centile, usually around the age of five years, or sooner if the girl is unusually short, e.g. when IUGR is a feature. The addition of Oxandrolone 0.05 mg/kg per day (maximum dose 2.5 mg daily) from the age of nine years showed a significant improvement in adult height over placebo in a UK study (Gault et al. 2011). Side effects were not reported with the dose regimen used but virilization has occurred in other studies using larger doses. Since Oxandrolone is not readily available in Europe, it tends to be reserved for girls aged >9 years in whom growth rate on GH is disappointing despite good compliance, as reflected by high/normal IGF‐1 levels. About 90% of girls with Turner syndrome require pubertal induction and the various oral and transdermal regimens are discussed in Chapter 5. The more physiological transdermal regimen might be expected to result in better pubertal growth but this hypothesis remains untested. In theory, delaying pubertal induction in Turner syndrome might be expected to increase final height. However, the UK Turner study, which randomized girls to receive pubertal induction at 12 or 14 years, found only marginal final height benefit from later induction. Moreover, late induction of puberty in Turner syndrome is now recognized to be undesirable both psychologically and in terms of uterine and bone health. It is now agreed that induction should usually begin at 11–12 years. However, the option of starting induction later in late‐diagnosed girls, to allow further time for growth, should be discussed with the patient and family. The strategy of introducing ultra‐low dose oestrogen from five years of age to promote growth (as well as bone, uterine, and cardiovascular health) is currently being investigated. GH therapy has been used to increase adult height in this condition. To date, robust adult height data are lacking (Giacomozzi et al. 2015). Growth impairment is relatively modest in many patients and in this situation the patient and family may not desire intervention. We recommend reserving GH use for patients with marked short stature (e.g. >‐2.5 SD) after discussion with the family. If GH is used to treat children and adolescents with Noonan syndrome, cardiac and skeletal surveillance should be enhanced. GH therapy has been shown to cause a dose‐dependent increase in height velocity in short patients with a history of SGA. High‐dose therapy for a period of two years induces catch‐up growth (De Zegher et al. 1998) and a European licence was granted for the treatment of short SGA children in 2003. The recommended dose varies widely, from 35 μg/kg/day in many European centres to an FDA‐approved dose of 67–69 μg/kg/day. However, evaluation of the safety and efficacy of GH therapy in SGA patients is challenging. The SGA population is heterogeneous and not all patients will have proven IUGR. Growth in response to GH may be restricted by an unusually rapid pubertal growth phase. Final adult height data are still limited and long‐term follow‐up data on cardiac and metabolic health are needed to evaluate safety, particularly with higher doses of GH. We recommend careful selection of patients who might benefit from treatment, comprising those with clinical evidence to suggest IUGR rather than SGA, and in whom height by the age of four years is < ‐2SD without catch‐up growth. We suggest a starting GH dose of 35 μg/kg/day, modifying this according to growth response, serum IGF‐1 levels, and bone age. Once a decision has been taken to treat with GH, this should be continued until final height is achieved, provided that the family are compliant, since discontinuous treatment is associated with catch‐down growth. Chronic renal failure is a potent cause of poor growth (Tonshoff and Mehls 1999). Treatment with GH has been extensively studied but this modality is used less often now with the advent of effective strategies which include: GH is occasionally used post‐transplantation, but better graft outcomes and reduced steroid use have decreased its role. Since patients are partially GH‐resistant, pharmacological doses of 50 μg/kg/day are required if GH is to be prescribed. The use of GH in renal disease should be monitored in a specialist renal department with experience of this treatment. Patients with skeletal dysplasia have normal GH secretion. GH can improve short‐term height velocity in achondroplasia with a typical first ‐ear response of 2–3 cm (Hagenäs and Hertel 2003). However, the benefit in terms of quality of life (e.g. being able to reach objects, self‐dress, etc.) are not well documented and there are no satisfactory data on the final height benefit of continuous treatment. One approach is to give GH in childhood to optimize linear growth, followed by limb lengthening from 13 years of age. However, non‐intervention is chosen by many families. The optimal dose is not known, but will be greater than for classic GH deficiency. We do not recommend the routine use of GH in this group of disorders. Short stature from chronic illness is best managed by treating the primary illness itself. This is dramatically demonstrated in Crohn disease, where successful resection of the inflamed bowel results in spectacular catch‐up growth (Figure 3.12). It is important to optimize nutrition and minimize steroid dosage. Newer treatments including anti‐TNFα therapy in Crohn disease and juvenile idiopathic arthritis (JIA) have led to significant improvements in growth. Treatment with GH is reserved for patients in whom growth remains poor despite every aspect of the chronic illness being managed as well as possible. Use of high dose recombinant GH in Crohn disease and JIA significantly increases growth rate but not completely, suggesting a degree of resistance at the level of the growth plate. Long‐term data on the benefit of GH therapy for adult height is awaited and ideally patients should only be treated in the context of a prospective clinical trial. Figure 3.12 Increase in height velocity following successful resection of inflamed bowel in prepubertal and early pubertal patients with Crohn disease. Source: After Walker‐Smith (1996). Induction of puberty is indicated in boys aged >14 years and girls aged >13 years with no pubertal development and earlier in selected cases when pubertal progress is slow or when delayed maturation is causing distress to the patient. Psychosocial deprivation may also be regarded as a chronic disease state. The best therapy for associated growth failure is to correct or reverse the disadvantageous home setting or, if this is not possible, to remove the child to a supportive environment. GH therapy is not indicated in this context nor is it effective. Recombinant IGF‐I was developed in the late 1980s and is the only therapy available for treatment of severe genetic GH resistance. It is effective in stimulating growth in Laron syndrome. However, a more common therapeutic indication for IGF‐I is still to be found. A compound which complexes IGF‐I with IGFBP‐3 has also been developed. This physiological approach is logical; however, its clinical efficacy for treatment of growth disorders remains to be demonstrated. Gonadotrophin‐releasing hormone (GnRH) analogue therapy has been used in conjunction with GH. The rationale of this approach is to suppress gonadotrophin secretion and arrest bone maturation, thus allowing a longer period for GH treatment. A prospective study with GnRH in 21 GH‐treated subjects reported an increase in adult height SD (Mericq et al. 2000). This strategy has not found widespread acceptance. However, the combination of early puberty and GH insufficiency, for example after cranial irradiation, constitutes a mandatory indication for combination treatment using GH and GnRH analogue to optimize growth before epiphysial closure. In this situation, puberty should be allowed to continue when an appropriate age is reached, continuing with GH therapy. Controversially, a combination of GH and GnRH analogue can be used to increase adult height in children with simple virilizing congenital adrenal hyperplasia when linear growth is poor despite minimal suppressive treatment with glucocorticoids. It is now well established that patients with severe GH deficiency, especially if accompanied by other anterior pituitary hormone deficiencies, require adult follow‐up to pre‐empt and treat the consequences of adult GH deficiency which include incomplete accretion and/or loss of bone mass with increased fracture risk, decrease in muscle mass, and increased fat mass (particularly visceral fat), disturbance of insulin and lipid metabolism, and increase in cardiovascular morbidity. Patients with the adult GH deficiency syndrome may experience marked fatigue with reduction in strength and exercise tolerance, and hence reduced quality of life. For this reason, it is essential to organize transitional care to an adult endocrine clinic for GH‐deficient adolescents leaving paediatric care (Clayton et al. 2005). The challenge is to convince the patient that this is necessary and beneficial, given that teenagers often view hospital attendance as tedious and non‐essential. Experience has shown that the setting up of a transition clinic in the paediatric centre, with quarterly joint clinics attended by the paediatric and adult endocrinologists increases the likelihood of successful long‐term follow‐up. Girls with Turner syndrome require transfer to a gynaecologist or adult endocrinologist, preferably in the context of an adult Turner clinic for surveillance of their reproductive, thyroid, bone, cardiac, and ENT/hearing status. If an adult Turner clinic is created then whoever runs it, whether reproductive endocrinologist, gynaecologist, or adult endocrine physician, needs to liaise closely with other disciplines, particularly cardiac and ENT.
Short Stature
Introduction
Definitions of short stature, failure to thrive, and growth failure
Physiology of growth
Normal linear growth
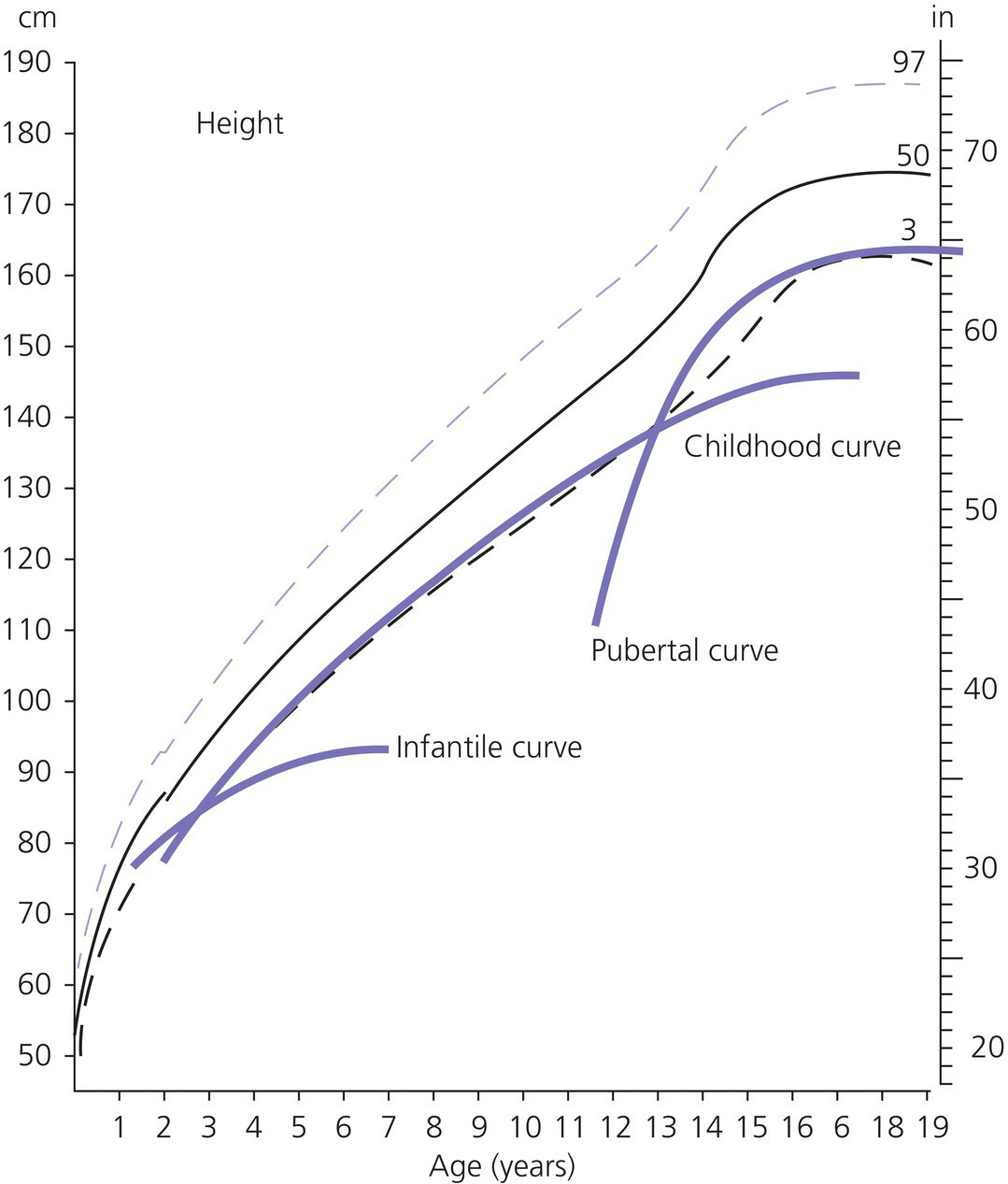
Infantile phase
Childhood
Puberty
Endocrine control of growth
GH secretion
The growth hormone‐insulin‐like growth factor (GH‐IGF) axis
Growth at puberty: the effect of sex hormones on the GH‐IGF axis and growth plate
Clinical assessment of growth
Techniques of measurement
Weight
Measurement of standing height
Measurement of supine length in children from birth to two years of age
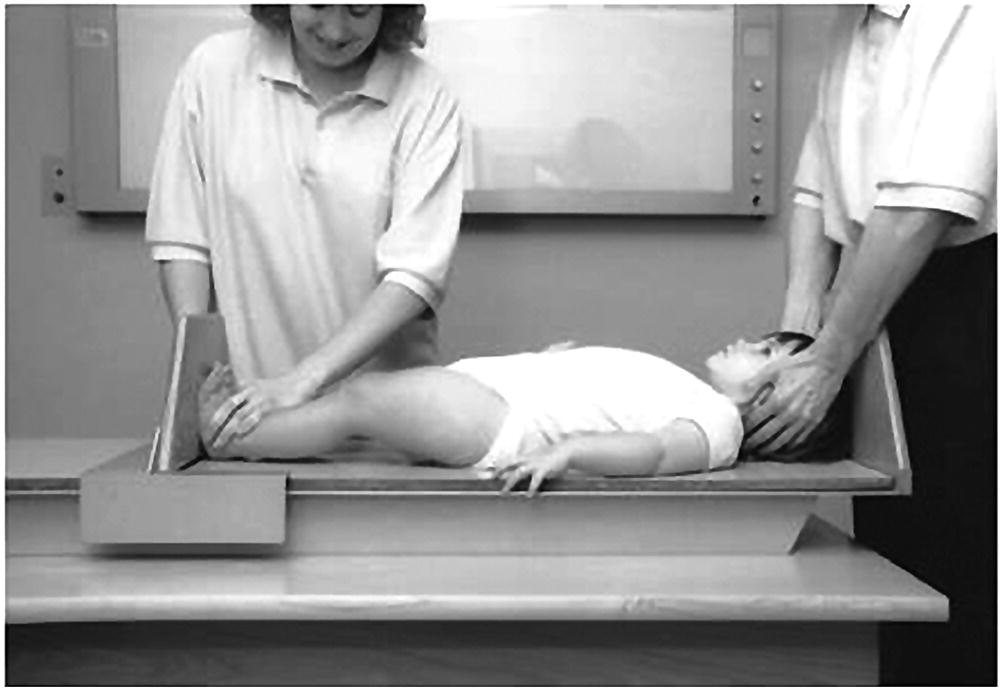
Sitting height
Upper arm circumference
Head circumference
Reliability
Decimal age
Calculation of height velocity
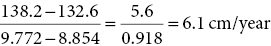
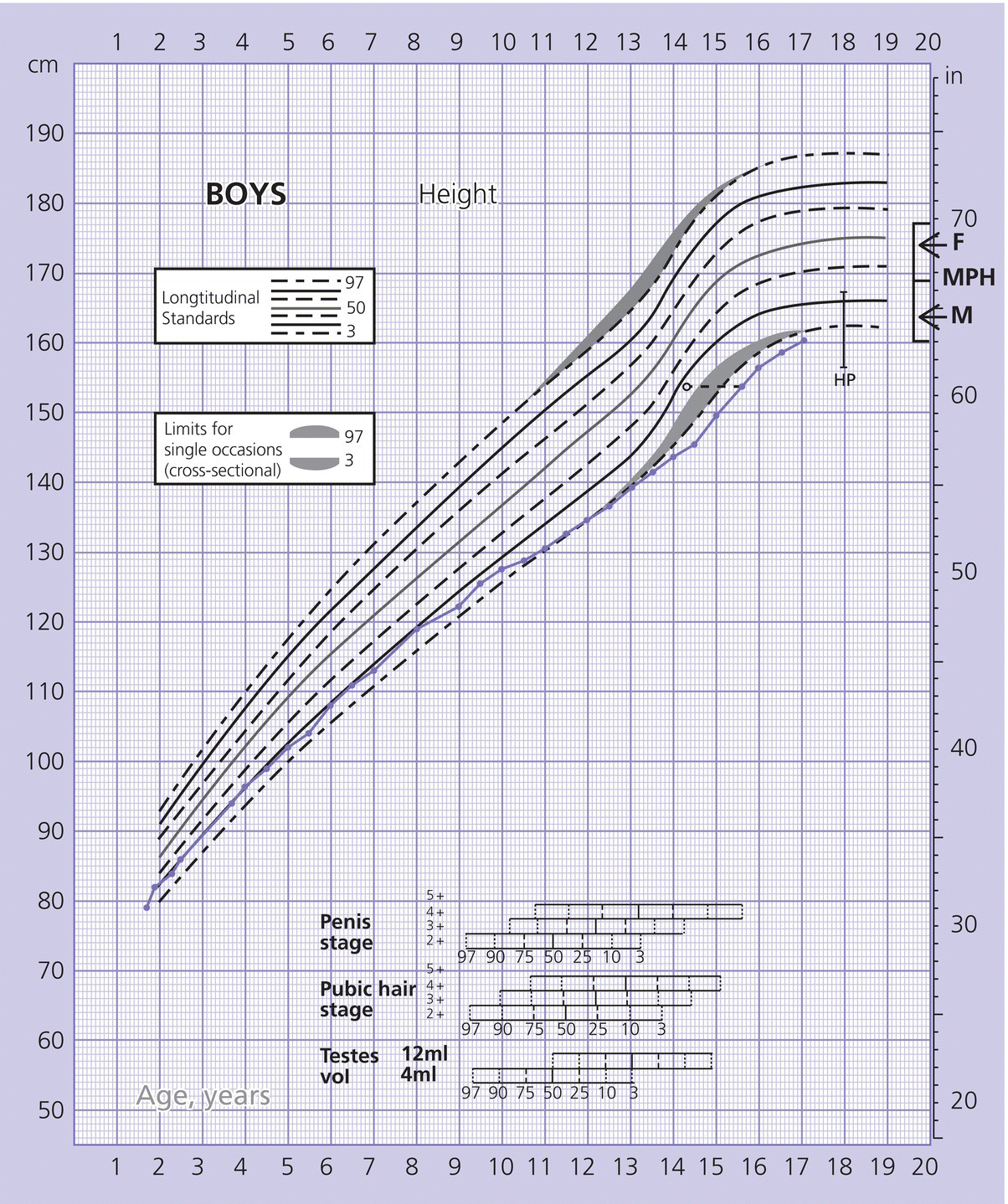
Parental height and target range
Pubertal staging
Height and height velocity standard deviation score
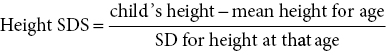
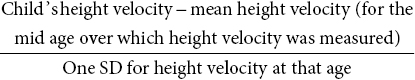
Body mass index
Assessment of skeletal maturity
The Greulich and Pyle method
The Tanner–Whitehouse method
Automated bone age assessment
Growth charts
Clinical assessment of short stature
Auxology
History
Examination (with child standing initially)
Clinical diagnosis
Investigation of short stature
Marked short stature (Ht SDS < −2.5)
Height centile below parental target range centiles
Abnormal/inappropriate height velocity for stage of maturity
History suspicious for chronic disease
History of neonatal hypoglycaemia and prolonged jaundice (suggestive of hypopituitarism)
Dysmorphic features suggestive of underlying syndrome (e.g. Turner and Noonan syndromes)
Pubertal delay
Extreme parental concern
Laboratory investigations for short stature
X‐ray of left wrist and hand for skeletal maturity (‘bone age’)
Full blood count, ESR
Creatinine, urea, electrolytes
Calcium, phosphate, liver function tests
Ferritin, tissue transglutaminase (TTG) (for coeliac disease)
Karyotype
T4, TSH
IGF‐I, cortisol, prolactin
Skeletal survey if dysplasia suspected
Differential diagnosis of short stature
Normal short stature
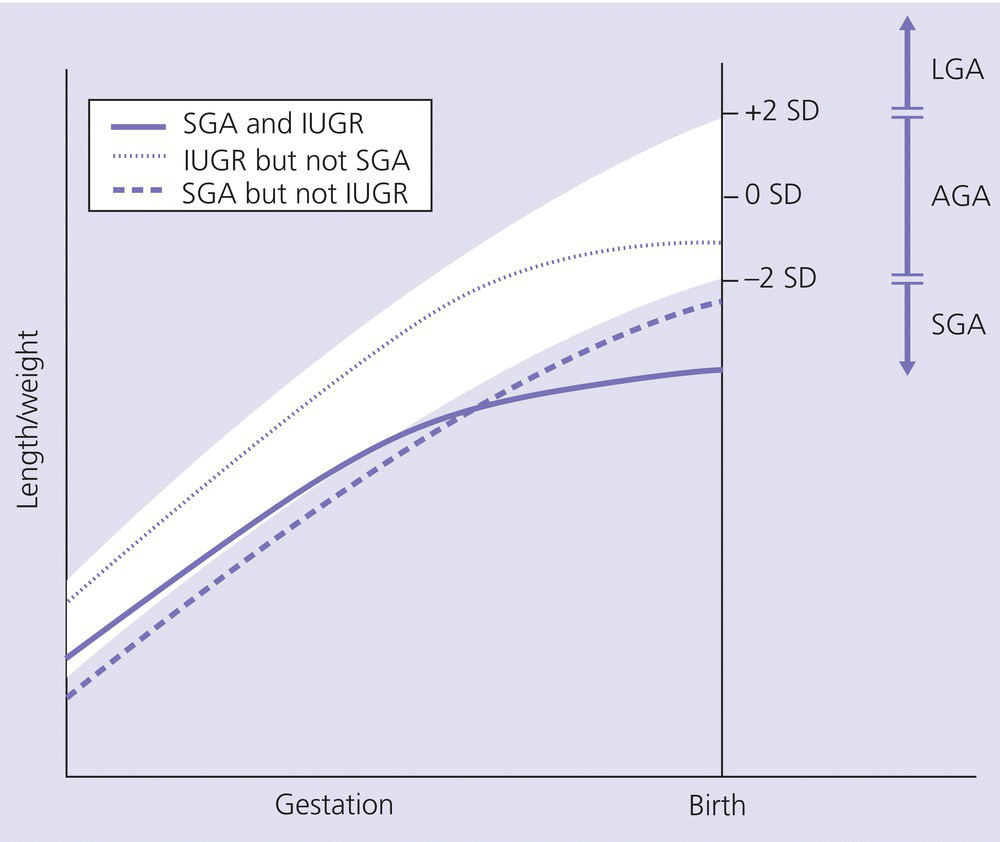
Short stature following smallness for gestational age (SGA)
Dysmorphic syndromes
Skeletal dysplasias (e.g. hypochondroplasia)
Chronic disease (e.g. coeliac disease, Crohn disease, cystic fibrosis)
Psychosocial deprivation
Endocrine disorders
Other
Normal short stature
Familial/Genetic short stature
Constitutional growth delay
Short stature related to smallness‐for‐gestational age and intrauterine growth restriction
Dysmorphic syndromes
Russell‐silver syndrome
Foetal alcohol spectrum disorder (FASD)
Turner syndrome
Features attributable to skeletal dysplasia
Ovarian dysgenesis
Chronic middle ear disease
Lymphoedema‐related
CNS‐related
Systemic malformations and problems
Immune and inflammatory disease (e.g. Hashimoto’s thyroiditis, coeliac and inflammatory bowel disease)
Miscellaneous – naevi, ptosis, epicanthic folds, oblique palpebral fissures, low set/rotated ears, low hairline 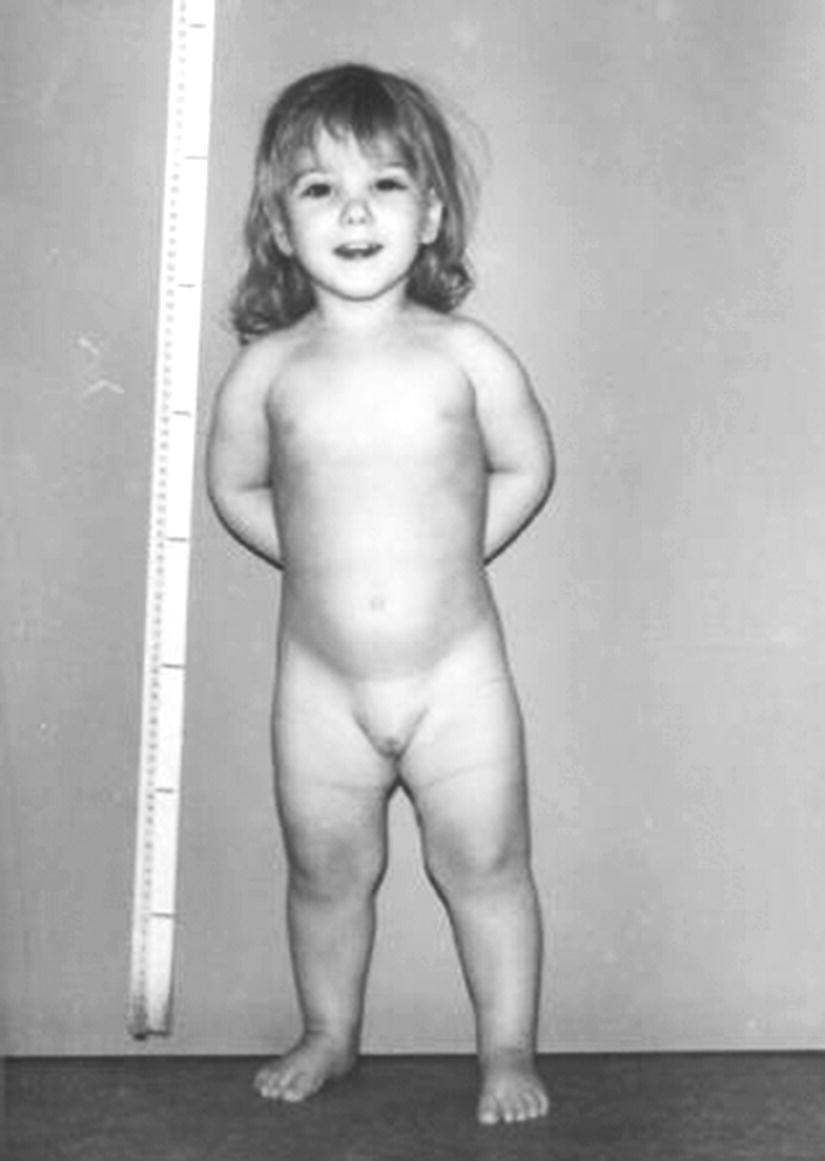
Genetic aspects
Growth (see Appendix 2)
Diagnosis and follow‐up
Noonan syndrome
Characteristic facies
Skeletal problems with:
Cardiac defects (usually pulmonary stenosis; also atrial septal defect, hypertrophic cardiomyopathy)
Gonadal problems (cryptorchidism in most males)
Delayed puberty
Lymphoedema (neck webbing)
Mild educational difficulties
Coagulation defect causing bleeding tendency 
Williams syndrome
Aarskog syndrome
Skeletal dysplasias
Achondroplasia
Activating mutation of fibroblast growth factor receptor 3 (FGFR3) gene (located on 4p16.3) in 99% of cases
Usually de novo mutation; autosomal dominant transmission
Normal intelligence
Severe shortening of long bones
Relatively long trunk with lumbar lordosis
Large head with hypoplastic mid‐face
Hypotonia and ligamentous laxity with associated limb pains
Narrow spinal canal with risk of symptoms from cord compression
Childhood height − 5 to – 6 SDS
Adult height approximately 132 cm in males, 125 cm in females
Hypochondroplasia
Activating FGFR3 mutation in 70% of cases
Autosomal dominant with variable severity
Normal head and face
Mild disproportion with short limbs
Short, broad hands and feet
Characteristic X‐ray changes with failure of interpedicular distance to increase in a cephalocaudal direction
Blunted pubertal growth spurt
Approximate adult height 155 cm in males, 142 cm in females
Spondylo‐epiphyseal‐dysplasia (SED)
Group of conditions including SED congenita due to mutations in the COL2A1 gene encoding type II collagen (autosomal dominant); and SED tarda which can be X‐linked (mutations in SEDL gene), dominant or recessive
Shortening of trunk and limbs with disproportionately short back
Relatively normal‐sized hands and feet
Atlanto–axial instability
SED congenita
Associated with myopia and hearing loss
Pain, especially in hips and back
Thoracic kyphosis and barrel chest
Coxa vara with waddling gait
SED tarda
Milder form with later presentation
Chronic disease
Psychosocial deprivation
Endocrine disorders
GH insufficiency and deficiency
Definition and cut‐offs

Severe GH deficiency
Presents before age three years (unless acquired)
Obvious short stature
Subnormal height velocity from birth, becoming more abnormal with age
Neonatal hypoglycaemia and hyperbilirubinaemia
Micropenis
Possible associated anterior pituitary hormone deficiencies (TSH, ACTH, LH, FSH)
Excess subcutaneous fat, increasing with age
Mid‐facial hypoplasia (only in extreme cases)
Possible features of septo‐optic dysplasia
Delayed skeletal maturation
Maximum stimulated GH concentration < 3 μg/L
Mild GH deficiency
Unlikely to present before school entry
Less severe short stature
Subnormal height velocity documented by careful auxology over minimum interval of 12 months
Isolated GH insufficiency
Normal subcutaneous fat
Delayed skeletal maturation
Delayed puberty
Maximum stimulated GH concentration 3–7 μg/L (3–10 μg/L in some centres)
Transient GH insufficiency: Conditions in which GH levels may be <7 μg/L on initial stimulation testing but normal on retesting
Permanent GH deficiency: Conditions in which GH levels are < 7 μg/L due to impairment of hypothalamo–pituitary axis
Congenital
Inherited causes
Structural defects
Hypothalamic disorders
Acquired
Tumours adjacent to hypothalamo–pituitary axis
Head injury
Surgery to the H‐P axis
Cranial radiotherapy (e.g. for medulloblastoma)
Granulomatous disease
Isolated GH insufficiency and deficiency
Idiopathic isolated GH insufficiency
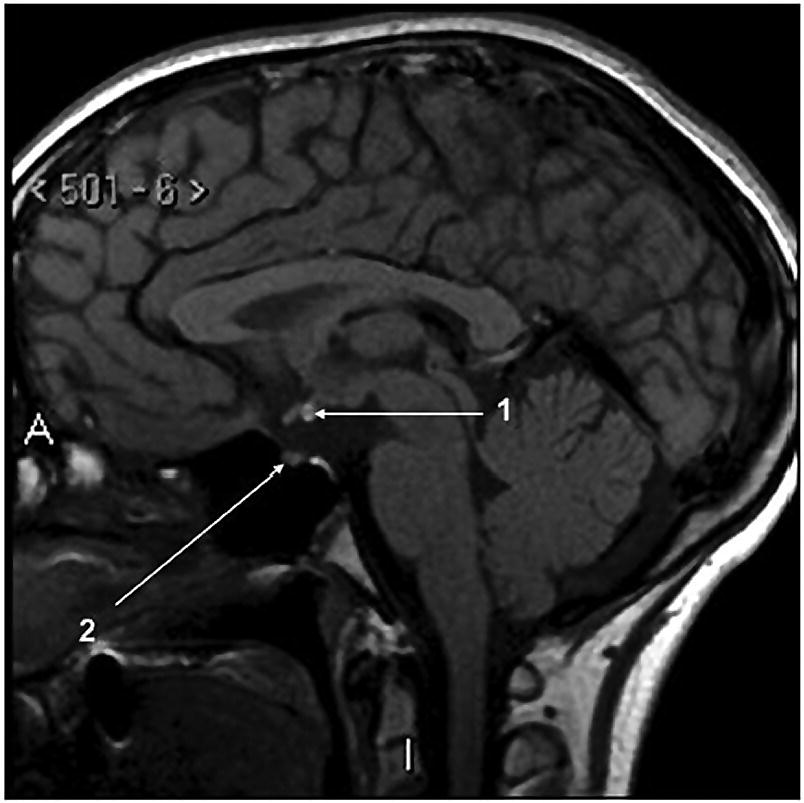
Multiple anterior pituitary hormone deficiencies
Idiopathic multiple anterior pituitary hormone deficiencies
Genetic causes of multiple anterior pituitary hormone deficiencies
Congenital structural CNS defects
Acquired causes of multiple anterior pituitary deficiencies
CNS tumours
Craniopharyngioma
Germinoma
Optic nerve glioma
Histiocytosis
Cranial irradiation
Diagnosis of GH insufficiency and deficiency
Auxology
Biochemical diagnosis
Physiological tests
Pharmacological tests
Glucagon test
Dose
15 μg /kg intramuscularly
Sampling
GH, cortisol, glucose at 0, 30, 60, 90, 120, 150, 180 min.
Complication
Hypoglycaemia, particularly in young children; nausea
Requirement
Doctor or specialist nurse in attendance throughout test
Contraindication
Epilepsy in young children
Clonidine test
Dose
0.15 mg/m2 orally
Sampling
GH at 0, 30, 60, 90, 120, 150, 180 min.
Complication
Hypotension
Requirement
BP monitoring
Insulin‐tolerance test
Dose
0.15 units/kg intravenously
Sampling
GH, cortisol, glucose at 0, 20, 30, 60, 90, 120 min.
Complication
Hypoglycaemia
Requirement
Doctor or specialist nurse at bedside throughout test, blood glucose <2.2 mmol/
Contraindication
Age < 5 years, epilepsy
Arginine test
Dose
0.5 g kg−1 intravenously over 30 min
Sampling
GH at 0, 30, 60, 90, 120, 150 min.
Complication
Nausea; irritation at iv site
Sex steroid priming prior to GH stimulation testing
Choice of stimulation tests
Execution of GH stimulation testing
Other endocrine causes of short stature
Hypothyroidism
Cushing’s syndrome
GH resistance
Idiopathic short stature (ISS)
Treatment of short stature
Constitutional delay of growth and puberty
Short stature
Low height velocity (<4 cm yr−1)
Delayed secondary sexual development
Abnormal body proportions (long legs, short trunk)
Reduced bone mineral density
Psychological distress related to:
Parental concern
Aims of treatment
Males
Females
GH insufficiency
Early diagnosis and initiation of therapy; central role of the endocrine specialist nurse
Dose 25–30 μg/kg/day (5 mg/m2/week) given subcutaneously in the evening, 7 days a week
4–6 monthly clinic visits for assessment including auxology and pubertal staging
Enquiry as to the number of injections missed between visits
Input from endocrine specialist nurse if growth response disappointing
6–12 monthly IGF‐I measurement to monitor dosage and compliance
Annual bone age
Discontinuation of GH at completion of growth (height velocity < 2 cm/year)
Retesting of GH status prior to adult transfer (IGF‐I and stimulated GH levels)
Transition clinic for adult transfer if permanent GH deficiency confirmed
Adverse events
GH therapy in non‐GH‐deficient disorders
Idiopathic short stature
Turner syndrome
Noonan syndrome
Short stature associated with SGA
Renal disease
Skeletal dysplasias
Chronic paediatric diseases
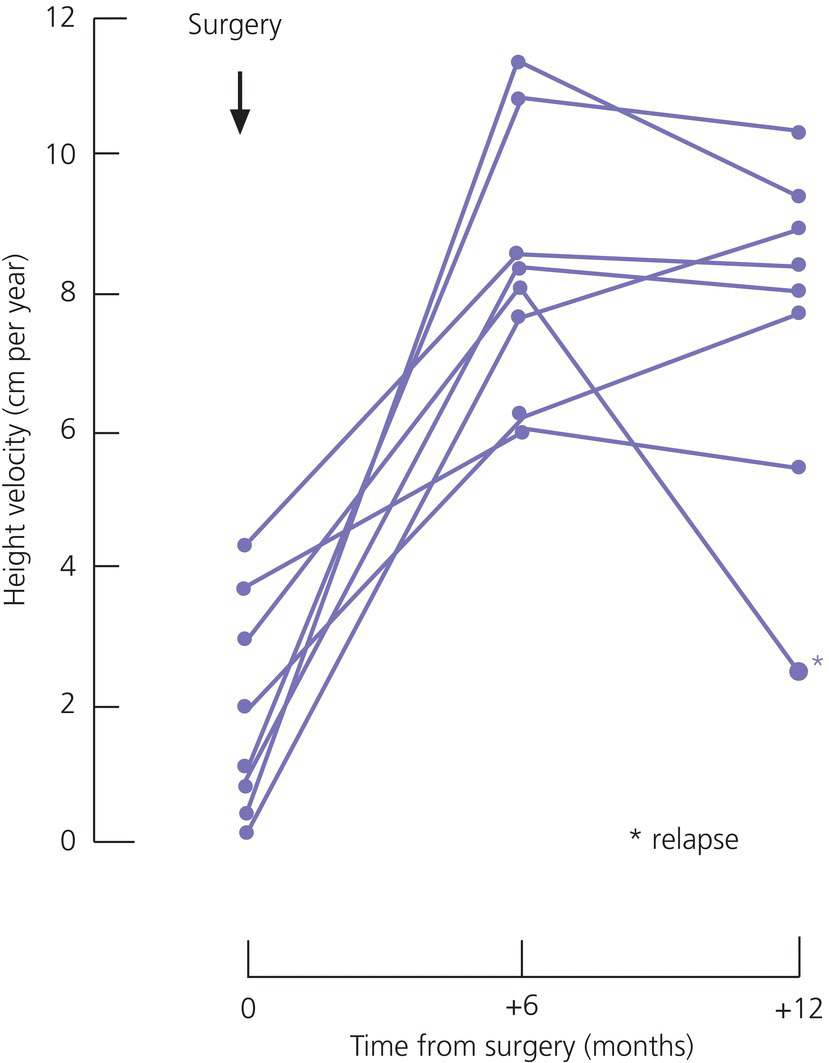
Psychosocial deprivation
IGF‐I
Use of gonadotrophin‐releasing hormone (GnRH) analogue therapy to improve growth in GH‐treated patients
Transition
When to involve a specialist centre
Future developments
Controversial points
Potential pitfalls
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


