Fig. 10.1
In cell culture systems, cellular senescence is described as a decrease in proliferative capacity and eventual cessation of proliferation with increasing number of population doublings (PDs). This property of normal cells is also described as finite life span or Hayflick limit in the literature. Tumor-derived and stem cells contain telomerase activity and/or maintain telomere length. Such cells are immortal and do not exhibit cellular senescence in culture.
10.1.1 Cellular and Molecular Features of Cellular Senescence
Cellular senescence traditionally has been confused with cell death. Contrary to cell death, senescent cells are metabolically active and resistant to apoptosis [3, 4]. Initial characterization of senescent phenotype in culture from various laboratories suggested that senescent cells stop synthesizing DNA as determined by tritium-labeled (3H) Thymidine or 5-bromo-2′-deoxyuridine (BrdU) incorporation, and are irreversibly arrested with a G1 DNA content [5]. The morphological features of senescent cells include big flat and vacuolated appearance [5]. Senescent cells express a senescence-associated β-galactosidase (SA-β-gal) marker, which is biochemically detected using X-gal as a substrate at pH 6.0 in human cells [6]. Increase in this enzyme activity probably reflects an increase in lysosomal compartment of senescent cells [7, 8]. The marker offers simple yet powerful tool to identify senescent cells in culture and in vivo, and has been widely used in several cancer-related studies.
The molecular features of senescent cells include altered expression of several genes [5, 9]. For example, several proliferation-associated genes such as gene encoding c-Fos, c-Myc, cyclin A, cyclin B, polo kinase PLK, centromere-associated protein CENP-A and CENP-F, and c-Myb and c-Myc are expressed, but downregulated in senescent fibroblasts [5, 9]. On the other hand, genes which encode proteins associated with maintenance and remodeling of ECM (extracellular matrix), such as stromelysin 1, stromelysin 2, collagenase, gelatinase, human macrophage metalloproteinase (HME), tPA, uPA, PAI 1, PAI 2, cystatin M, thrombospondin, dermatopontin, fibromodulin, collagens VI and XV, elafin, cathepsin D, cathepsin L, serpin b2, and other ECM-associated proteins are upregulated in senescent fibroblasts [2, 9].
10.1.2 Types of Cellular Senescence
The senescence phenotype which Hayflick and Moorehead first described is now known as replicative senescence. Replicative senescence is caused by repeated cell division or mitoses (Fig. 10.2). The prime cause of replicative senescence is telomere shortening or dysfunction [10]. However, it is very well established that a similar phenotype also results due to non-telomeric signals such as stress signals (Fig. 10.2) [10]. For example, cellular senescence in cultured murine cells is thought to be due to stress induced by culture conditions [11]. In addition, oncogenic and mitogenic signals, such as strong oncogenic signal provided by activated H-RAS can induce senescence in primary cells (Fig. 10.2). Thus, senescence inducing signals can be telomeric or non-telomeric (Fig. 10.2). The non-telomeric signals include oncogenic, mitogenic, DNA damage, and undefined stress signals (Fig. 10.2). Senescence induced by non-telomeric signals is termed accelerated senescence, premature senescence, stress or aberrant signaling induced senescence (STASIS), and extrinsic senescence [12, 13].
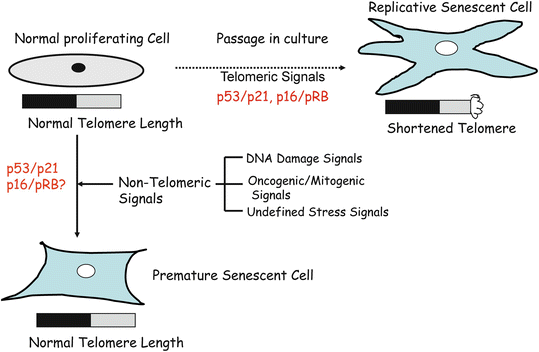
Fig. 10.2
Normal human cells undergo two types of senescence-replicative and premature senescence. Replicative senescence is caused by cell divisions (mitoses); during each cell division, telomeres shortens about 50–100 bp (base pairs). When telomeres length gets critically short to 1–2 kb (kilo base) from 8–12 kb, cells stop dividing and acquire a distinctive morphology. On the other hand, premature or accelerated senescence is caused by non-telomeric signals such as DNA damaging agents, constitutive oncogenic/mitogenic signaling, and undefined stress signals. During premature senescence, telomere length remains unchanged; however, the distinctive senescent morphology is still acquired by the cells.
10.1.3 Mechanisms of Cellular Senescence
A large number of telomere binding proteins cap the end of chromosome known as telomeres, which consist of long stretch of repetitive sequences, such as (TTAGGG) n in humans [14]. In human somatic cells, progressive telomere shortening or telomere dysfunction appears to be the primary cause of cellular senescence [14, 15]. In most cases, the enzyme telomerase and telomere associated proteins maintain telomere homeostasis and telomere length. The catalytic subunit of telomerase (TERT) together with its RNA component (TERC), builds telomere repeats at the chromosome ends, which otherwise, owing to asymmetric DNA replication, are progressively lost [14].
Most human somatic cells do not contain sufficient telomerase to maintain telomere length [16], resulting in telomere shortening after each round of cell division. Consistent with this finding, exogenous expression of telomerase can either increase or stabilize telomere length in normal human cells, and in some cases results in cell immortalization [17]. It is thought that telomere dysfunction or uncapping triggers a DNA damage response, which activates p53 pathway resulting in proliferation arrest [18–20]. Consistent with this model, senescent cells are enriched in the nuclear foci of phosphorylated histone H2AX (γ-H2AX), p53 binding protein 53BP1, NBS1, the phosphoS966 form of SMC1 and MDC1, and inactivation of CHK1 and CHK2 (checkpoint kinases) can restore S phase progression in senescent cells [18–20]. Oncogene-induced senescence (OIS) has also been shown to be a DNA-damage checkpoint-response mediated by ATM and CHK2 [21, 22].
The constitutive activation of p53 via ATM pathway results in induction of p53 target genes most notably p21 [23], which inhibits the activity of CDK2/cyclin E kinase. Inhibition of CDK2/cyclin E kinase activity results in hypophosphorylation of pRB (retinoblastoma tumor suppressor), which very likely mediates cell cycle arrest during senescence (Fig. 10.3). In addition to p21, other growth inhibitory p53 target genes may also play a role in cellular senescence [10]. Due to p53 and p21 activation and resulting G1 arrest, many proliferation-associated genes such as CDK1, CDK2, cyclin A, cyclin B, and E2F1 remain repressed in senescent cells. In a parallel pathway, p16INK4a (CDKN2A) is upregulated during senescence [10]. p16INK4a inhibits the activity of other CDK-cyclin kinases such as CDK4-cyclin D1 and CDK6-cyclin D1. CDK4, CDK6, and cyclin D are expressed in senescent cells, but the activity of CDK-cyclin D complexes is lost during senescence. Thus, the repression of CDK kinases via multiple mechanisms results in hypophosphorylation of pRB. The hypophosphorylated or unphosphorylated pRB maintains permanent growth arrest phenotype of senescent cells (Fig. 10.3) [10]. Expression of p16INK4a is negatively regulated by BMI-1 oncoprotein [13], while p53 is negatively regulated by MDM2, PIRH2, COP1, and ARF-BP1 [24]. These regulators of p16INK4a and p53 are likely to impact cellular senescence and replicative life span of cells in vitro and in vivo (Fig. 10.3).
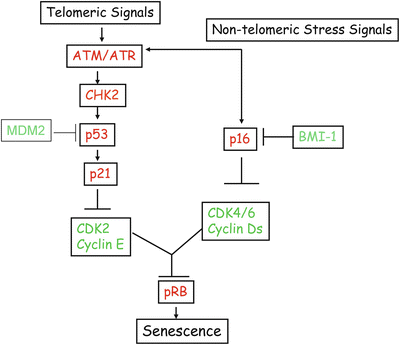
Fig. 10.3
Telomeric and non-telomeric stress signals induce senescence via tumor suppressor pathways. These signals activate a DNA damage response pathway mediated by ATM and CHK2. These DNA damage inducible kinases activate p53, which via p21 suppresses the CDK2/cyclin E activity to maintain pRB in unphosphorylated or hypophosphorylated (growth inhibitory) state. Stress signals in culture also activate p16INK4a (independent of p53). Very little is known about the upstream signaling molecules that mediate p16INK4a induction. p16 primarily inhibits CDK4/6 cyclin D activity. p16INk4a together with p21 and possibly other CDK inhibitors maintain pRB in a permanent growth inhibitory state, which ensures stability and irreversibility of senescent phenotype. p16INK4a is negatively regulated at the transcription level by polycomb proteins, in particular BMI-1. p53 is negatively regulated at the protein level by MDM2 and other ubiquitin ligases.
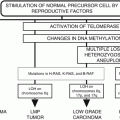
10.1.4 Telomere-Dependent Senescence and Cancer
Telomeres and Telomerase expression are linked to cancer in many ways. First, more than 90 % of tumors express telomerase [25] and tumor-derived cells usually maintain telomere lengths of their chromosomes. Second, several in vitro studies have shown that telomere attrition can induce senescence or apoptosis in tumor cells [26]. Third, the ectopic expression of telomerase in somatic cells prevents telomere shortening and in some cases results in immortalization of cells [17, 26], which may increase the possibility of accumulation of additional genetic abnormalities resulting in cancer initiation and progression. Indeed several studies have shown that hTERT overexpression is one of the four pivotal steps required to convert a normal human somatic cell into a cancer cell [27, 28]. These studies also showed that bypass of senescence by inactivating pRB and p53 is one of the other four requirements for in vitro transformation of human cells [10, 28, 29]. Based on these studies, one can hypothesize that telomere shortening should limit cancer progression in vivo. Until now, compelling evidence for such a hypothesis has been lacking because the laboratory mouse which is the most studied in vivo cancer model harbors very long telomeres.
Taking advantage of a mouse knockout model of telomerase (mTR −/−), which lacks the essential RNA subunit of the enzyme telomerase, Felder and Greider recently showed that indeed short telomeres limit tumor growth in vivo by inducing cellular senescence [30]. Felder and Greider crossed mTR −/− mice with Eμ-Myc transgenic mice, which develop very aggressive Myc-driven Burkitt’s lymphomas. It was found that at generation six (G-6), when the telomeres in mTR −/− mice get short enough, the progression of Eμ-Myc-induced Burkitt’s lymphomas was completely blocked by apoptosis. To negate apoptosis in these animals, the investigators used Bcl-2 overexpression, and showed that short telomeres still inhibited lymphoma formation in these animals despite inhibition of apoptosis. The staining for SA-β-gal marker, p16INK4a, and p15INK4b indicated that precancer cells in this setting were undergoing cellular senescence, implying that short telomere-driven senescence acts as a strong barrier to neoplasia in vivo [30]. Importantly, loss of p53 abrogated short telomere-induced senescence and promoted cancer progression [30]. Cosme-Blanco et al. also demonstrated that dysfunctional telomeres induce p53-dependent cellular senescence, which abrogated spontaneous tumorigenesis in mTR −/−p53 (R172P) (telomerase knockout with mutant p53 knockin) mouse model [31]. Thus, telomere-dependent senescence also acts as a strong barrier to tumor-progression in vivo.
If the telomerase expression is required for oncogenesis, then inhibitors of telomerase should be considered attractive cancer therapy reagents. Indeed, inhibitors of telomerase are known to induce senescence and/or apoptosis in cancer cells. For example, Damm et al. reported that a highly selective small molecule inhibitor of telomerase caused progressive telomere shortening and induced senescence in p53-positive as well as p53-negative cancer cell lines [32]. Similarly, synthetic telomerase inhibitors MST-312, MST-295, and MST-199 were reported to induce senescence in cancer cells [33]. Riou et al. also demonstrated that a series of G-quadruplex DNA ligands can inhibit telomerase activity, cause telomere shortening, and induce cellular senescence [34]. Several other inhibitors of telomerase such as BIBR1532 and GRN163L exhibit growth suppressive and anticancer activity [35]. These studies suggest that senescence induced by telomere shortening is a tumor suppressive mechanism, and compounds that inhibit telomerase activity and induce senescence can be used as cancer therapy reagents.
10.1.5 Oncogene-Induced Senescence and Cancer
In 1997, Serrano et al. first reported that oncogenic RAS did not transform primary human cells but instead invoked a phenotype similar to cellular senescence, suggesting that senescence can be prematurely activated in response to an oncogenic stimulus [36]. This phenomenon of premature senescence by oncogenes is now popularly known as OIS [37]. Apart from oncogenic RAS, OIS is known to be induced by RAF, MYC, E2F1, and constitutive MAPK signaling in normal cells [10]. Recently, MOS, cyclin E, and CDC6 oncogenes were also shown to cause OIS in human fibroblasts [21]. Since the discovery of Serrano et al. and others, OIS is now recognized as an important body defense mechanism against oncogenic insults. The in vivo evidence for such a role for OIS was reported in a series of papers published in 2005 [38–41]. It was found that the expression of B-RAF, K-RAS, and Eμ-N-RAS oncoproteins, or inactivation of the tumor suppressor PTEN induced a senescence response in vivo, and as a result, cancer progression was blocked at a very early stage [38–41]. Importantly, in these studies it was shown that cancer progression was only facilitated by additional mutations such as loss of p53, which caused bypass of senescence. Accordingly, malignant tumors that eventually arose in these models were largely devoid of biomarker of senescence including SA-β-gal.
Michaloglou et al. reported that benign nevi in human patients, which express oncogenic form of B-RAF, by in large are composed of senescent melanocytes as these cells express markers of senescence such as SA-β-gal and p16INK4a, while the malignant melanomas were devoid of these senescence markers [41]. In a transgenic model of lung cancer progression by K-RAS, it was shown that the premalignant tumors but not the malignant tumors contained senescent cells [40]. In the Braig et al. study, OIS was shown as an initial barrier in lymphoma development in Eμ-N-RAS transgenic mouse model [38]. It was shown that the deficiency of Suv39h1, a methyltransferase, which silences proliferative genes to induce senescence, is required for lymphomagenesis induced by Eμ-N-RAS transgene. Finally, Chen et al. showed that PTEN inactivation per se leads to induction of p53-dependent premature senescence in vitro and in vivo [39]. It was also shown that early stage benign human prostate cancer samples but not the late stage prostate cancer samples contained senescent cells [39]. Collectively, these studies strongly suggest that OIS acts as an initial barrier to cancer progression in vivo [42].
10.1.6 Senescence and Cancer Therapy
Both OIS and telomere-based senescence are important for cancer initiation and progression. Apart from cancer initiation and progression, senescence is also important for cancer treatment. Several recent studies have shown that chemotherapeutic drugs and radiation can induce senescence in cancer cells [43]. Induction of senescent phenotype as measured by the expression of SA-β-gal marker has been reported in a number of cancer cell lines by doxorubicin [44–46], cisplatin [47–49], hydroxyurea [50–52], camptothecin [53–55], etoposide [55], and other chemotherapeutic agents.
It has been suggested that DNA-interactive chemotherapeutic drugs such as doxorubicin, aphidicolin, cisplatin, and cytrabine induce a robust senescence-like growth arrest while other cytotoxic drugs, such as taxol and vincristine, induce it only minimally [56]. Apart from in vitro studies, recent studies provide compelling evidence that cellular senescence can be induced in vivo by chemotherapeutic drugs, and that senescence induction by chemotherapy drugs might be an important mechanism of cancer growth inhibition by these drugs [55, 57, 58]. te Poele et al. reported that 41 % of breast cancers from patients who had undergone a chemotherapy regimen stained positive for SA-β-gal marker and p16INK4a protein, while normal tissues adjacent to tumors were devoid of senescence markers [55]. The investigators concluded that like apoptosis, senescence is a p53-induced cellular response to chemotherapeutic drugs in vivo and that it is an important factor in determining treatment outcome [55]. The induction of premature senescence by chemotherapy treatment of human lung cancer was also recently demonstrated [57].
A study by Schmitt et al. provides clear evidence for the induction of senescence by cancer chemotherapy reagents in a mouse model [58]. The investigators generated Eμ-Myc-derived lymphomas in control-MSCV (vector), control-Bcl2, p53 null-MSCV (vector), and p53 null-Bcl2 mice. Mice bearing these four sets of lymphomas were treated with a chemotherapy drug CTX (cyclophosphamide). Using staining for SA-β-gal marker, p16INK4a, and p53, the investigators showed that CTX is able to engage a senescence program when apoptosis is inhibited by Bcl2 overexpression (control-Bcl2 set) [58]. Furthermore, as a result of senescence induction, control-Bcl2 lymphomas did not progress and these animals had a better prognosis [58]. This is in contrast to p53 null-MSCV and p53 null-Bcl2 lymphomas, which progressed rapidly [58]. Induction of a senescence-like phenotype has also been reported in rat mammary tumors undergoing treatment with chemopreventive agents [59]. Thus, chemotherapeutic drugs can induce a senescence-like stage in vivo and in vitro by upregulating p53 and/or p16INK4a, which may be an important mechanism of cancer growth inhibition by these drugs [10].
10.1.7 p53-Mediated Senescence in Cancer Treatment
p53 is mutated in a large number of human malignancies [60, 61]. p53 is also a major regulator of senescence and enforced expression of p53 or p21 in many cell types can induce a senescence-like phenotype [10]. Thus, it is natural to ask—does restoration of p53 in p53-deficient tumors induces senescence and stop cancer growth? Two elegant studies suggest that p53 restoration in a mouse model can cure cancer via induction of senescence and tumor clearance [62, 63]. In the first study, Xue et al. used RNA interference (RNAi) approach to conditionally regulate p53 expression in liver carcinoma in a mouse model [63]. The investigators showed that even a brief reactivation of endogenous p53 caused complete regression of the liver tumor. Importantly, the primary cause of cancer regression was not apoptosis, as one would have thought. Instead, cancer regression in this model involved induction of the cellular senescence program accompanied by the production of inflammatory cytokines [63]. The inflammatory cytokines triggered an innate immune response which enhanced tumor clearance. Thus, it was shown that p53-mediated induction of cellular senescence together with innate immune response can cure murine liver carcinomas [63].
Ventura et al. used a Cre–loxP-based conditional strategy to activate or inactivate endogenous p53 in a mouse model [62]. The investigators generated mice with a reactivatable p53 knockout allele by inserting a loxP flanked STOP cassette in the first intron of p53. This STOP cassette created a p53 null phenotype, which could be reversed to the p53-positive phenotype by removal of loxP sites (and STOP cassette) using a tamoxifen inducible Cre-recombinase. The p53-null mice in this setting developed lymphomas and sarcomas. After the tumors grew to a detectable size as measured by MRI imaging, the investigators restored p53 expression by administration of tamoxifen, which removed the STOP cassette and loxP sites. Within 24–48 h of activation of p53, it was shown that lymphomas regressed by apoptosis as determined by markers of cell death [62]. On the other hand, regression of sarcomas was only modest by 48 h and took several days for complete regression. The regressing sarcomas showed no apoptosis but rather were positive for markers of senescence such as SA-β-gal, p16INK4a, and p15INK4b. Thus, the investigators concluded that p53 restoration caused tumor regression in both lymphoma and sarcomas, although the outcome in the first case was based on apoptosis, and in the second case was based primarily on senescence induction [62]. The reasons for cancer clearance after senescence induction are not clear, but could involve immune response by the host.
10.2 Apoptosis and Cancer
Long before the role of senescence in pathogenesis of cancer was recognized, it was firmly believed that inhibition of apoptosis is an integral part of cancer development [64–66]. Current literature only reinforces this notion that apoptosis is an important determinant in the pathogenesis of cancer. Selective elimination of cancer cells by apoptosis is still the long sought promise of many cancer therapy drugs [67]. The process of apoptosis is irreversible and characterized by chromatin condensation, nuclear fragmentation, membrane blebbing and cell shrinkage, and formation of apoptotic bodies [68]. These properties of apoptotic cells are distinct and usually not associated with other forms of cell death such as necrosis, autophagy, and mitotic catastrophe [69]. In view of its highly regulated nature, apoptosis is also termed programmed cell death (PCD). Apoptosis is involved in many physiological processes such as embryonic development, morphogenesis and tissue homeostasis [70, 71]. It is dysregulated in many human diseases such as autoimmune diseases, AIDS, neurodegenerative diseases, and cancer [72]. Here, we briefly review the mechanisms of apoptosis, the role of apoptosis in cancer progression and induction of apoptosis by cancer therapeutics.
10.2.1 Mechanisms of Apoptosis
In mammalian cells, apoptosis is activated and executed through two major signaling pathways, the intrinsic and the extrinsic pathways (Fig. 10.4) [73, 74]. The intrinsic pathway is mitochondria-mediated and is triggered by intracellular cues such as DNA damage [73, 74]. The intracellular signals activated by DNA damage or other stresses result in the release of electron-transport chain intermediate Cytochrome-c (Cyt-c) to the cytoplasm from mitochondrial intermembrane space (IMS). Once released into the cytoplasm, Cyt-c facilitates the binding of procaspase-9 to APAF-1, which results in its activation and formation of the apoptosome containing activated caspase-9 [73, 74]. Release of Cyt-c from IMS is regulated by a balance between pro-apoptotic and anti-apoptotic members of the BCL2 (Bcl-2) family of proteins [74].
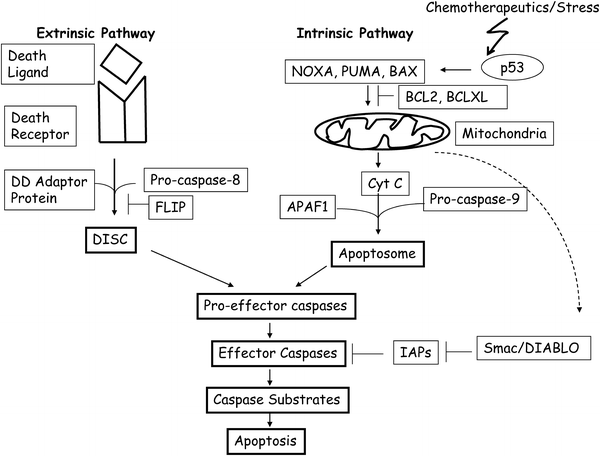
Fig. 10.4
A simplified cartoon of apoptotic pathways in mammalian cells. The extrinsic pathway of apoptosis is activated when a death ligand such as FASL, binds to pro-apoptotic receptor such as FAS. The downstream signaling is mediated by interaction of death ligand–death receptor complex with a death domain (DD) protein such as FADD. The association of these proteins with pro-caspase-8 results in DISC assembly, which activate effector caspases by proteolytic cleavage. Effector caspases cleave various caspase substrates to complete the process of apoptosis. FLIP inhibits DISC assembly by binding to caspase-8 and DD adaptor proteins. The intrinsic pathway of apoptosis is primarily mediated by mitochondrial release of Cyt-c. In response to DNA damage or other stresses, p53 is activated, which transcriptionally induce pro-apoptotic proteins such as BAX, NOXA, and PUMA. These proteins help release of Cyt-c from mitochondria. Release of Cyt-c is inhibited by BCL2 and BCLXL. Cyt-c release facilitates APAF1 and pro-caspase-9 assembly known as apoptosome, which activate casapase-9. Caspase-9 binds pro-effector caspases and activates them. IAPs negatively regulate effector caspases such as caspase-3, caspase-6, and caspase-7. Certain IAPs such as XIAP also binds and inhibits caspase-9. In response to apoptotic signals, mitochondria also releases an inhibitor of IAPs known as Smac/DIABLO. The role of these pro- and anti-apoptotic proteins is described in the main text.
The detailed mechanism of action of these pro-apoptotic and anti-apoptotic proteins is still not clear. It is thought that pro-apoptotic BH3 (BCL2 homology 3)-only proteins such as BIK, BAD, BIM, NOXA, PUMA, and multi-domain BH proteins such as BAX, BAK, BCLB, BLCG, and others increase the permeability of mitochondrial membrane by forming pores. On the other hand, anti-apoptotic BCL2 family members such as BCL2, BCLXL, BCLB, BCLW, and MCL1 prevent permeabilization of mitochondrial outer membrane by inhibiting the function of pro-apoptotic members of BCL2 family of proteins [74–76]. Several of these pro-apoptotic proteins such as NOXA, PUMA, and BAX are direct transcriptional targets of p53 [77–79], which is activated by DNA damage and other stress signals. p53 can also directly cause Cyt-c release from mitochondria and thus cause apoptosis without transcriptional activation of its target genes [80, 81]. Other molecules which regulate intrinsic pathway of apoptosis are IAPs (inhibitor of apoptosis) such as XIAP, which inhibits caspase-3, caspase-7, and caspase-9 [82]. XIAP in turn is inhibited by a pro-apoptotic protein Smac/DIABLO, which is released by mitochondria during apoptosis (Fig. 10.4) [83, 84].
The extrinsic pathway is death receptor-mediated and is activated at the cell surface by extracellular cues, whereby pro-apoptotic (death) ligand binds such as FASL, TNF, and TRAIL, bind and activate pro-apoptotic (death) receptors CD95/FAS, TNFR, and DR4/5 respectively [73, 74]. Death receptors contain related intracellular motifs known as death domains (DD), which interact with similar domains in corresponding adaptor molecules such as FADD and TRADD. Through the interaction of DD of death receptors and corresponding adaptors, DISC (death-inducing signaling complex) is assembled at the plasma membrane. Adaptor molecules also contain death effector domains (DED), which help recruit procaspase-8 via its own DED domain to DISC [73, 74]. Once at DISC, high local concentration of procaspase-8 leads to its activation via autoproteolytic cleavage. Subsequently, caspase-3, caspase-6, and caspase-7 are activated, which ultimately lead to apoptosis by cleaving their substrates (Fig. 10.4) [73, 74].
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


