SECONDARY HYPOGONADISM
Part of “CHAPTER 115 – MALE HYPOGONADISM“
HYPOGONADOTROPIC HYPOGONADISM
Hypogonadotropic hypogonadism may be caused by acquired or congenital defects. The presentation of the acquired forms of the disorder varies depending on whether the individual has gone through puberty and whether other anterior or posterior pituitary hormone deficiencies are involved. If the individual acquires the disorder before the onset of puberty, the presentation is that of a prepubertal male, as noted in Table 115-2. If puberty has occurred before the onset of the disorder, the presentation is with signs and symptoms of postpubertal testicular failure. The acquired forms of the disorder and some congenital forms have additional anterior pituitary deficiencies. If growth hormone is deficient, it is of clinical significance only if the individual is prepubertal and has not reached his adult height. After puberty, when the epiphyses of the long bones have fused and linear growth has ceased, growth hormone deficiency will no longer be apparent.
CLASSIC HYPOGONADOTROPIC EUNUCHOIDISM
Just as Klinefelter syndrome has become the prime example of hypergonadotropic hypogonadism, congenital hypogonadotropic eunuchoidism is the classic example of hypogonadotropic hypogonadism. As originally described by Kallmann and colleagues, this syndrome (Kallmann syndrome) is characterized by isolated gonadotropin deficiency and anosmia or hyposmia due to defective development of the olfactory bulbs.132,133 Other findings occasionally described with this disorder include midline cleft palate and lip, congenital deafness, cerebellar seizures, a short fourth metacarpal, and cardiac abnormalities.134,135
Kallmann syndrome is most commonly associated with autosomal dominant inheritance.134 The associated defects, especially anosmia, have enabled tracing of father-to-son transmission.136 However, kindreds have been reported that suggest an autosomal recessive or X-linked form of inheritance.137 The X-linked form of Kallmann syndrome is due to point mutations and exon deletions of the KAL gene located on the X chromosome at location Xp22.3.138,139 and 140 The gene consists of 14 exons and produces a 680-amino-acid, 76-kDa protein (anosmin-1) that can be glycosylated to form an 85-kDa protein product. The protein contains four fibronectin type III repeats associated with cellular adhesion functions, and a four-disulfide core motif that is commonly associated with antiprotease activity. The question of other forms of inheritance will be further defined as more men and women with the syndrome become fertile as a result of newer modes of therapy, and their progeny are studied.
The incidence of this syndrome is ˜1 in 10,000 male births.134 These men present as prepubertal eunuchs. They usually have no evidence of pubertal physical findings and their skeletal proportions are eunuchoid, with the ratio of upper body segment (pubis to vertex) to lower body segment (pubis to floor) decreased; the arm span is at least 6 cm greater than the height. These body proportions result from a failure of epiphyseal fusion and continued long bone growth. Beard and pubic hair growth are absent or minimal. Most patients retain their prepubertal subcutaneous body fat (Fig. 115-16). Hyposmia is present in most cases but may be missed unless specifically tested using appropriate olfactory sensation materials141 (Fig. 115-17). Muscle mass and strength remain at prepubertal levels. Although some CNS problems have been described,142 mental retardation is not one of them. A small phallus and temporal facial wrinkling caused by hypogonadism are common in this disorder. Gynecomastia may occur; peripheral aromatization of adrenal androgens may be contributory. Testicular development remains at prepubertal levels, with a few patients showing some testicular enlargement at puberty.143 However, unlike in Klinefelter syndrome, the testes are of normal prepubertal size because no tubular scarring or loss of germ cells is present. The Leydig cells are immature (as would be seen without LH stimulation), although normal numbers of interstitial cells are present, and the development into mature testosterone-producing Leydig cells occurs with gonadotropin stimulation (Fig. 115-18).
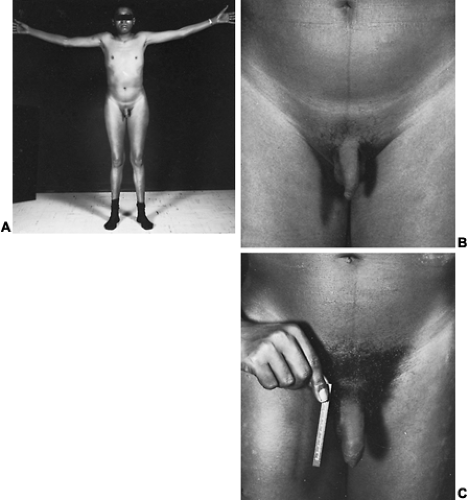 FIGURE 115-16. A, Patient with hypogonadotropic hypogonadism (Kallmann syndrome). Note the long legs and the increased arm span. Body hair is sparse, and the penis is small (B). C, Note the increase in penile size and pubic hair after 6 months of therapy with testosterone. |
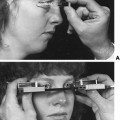
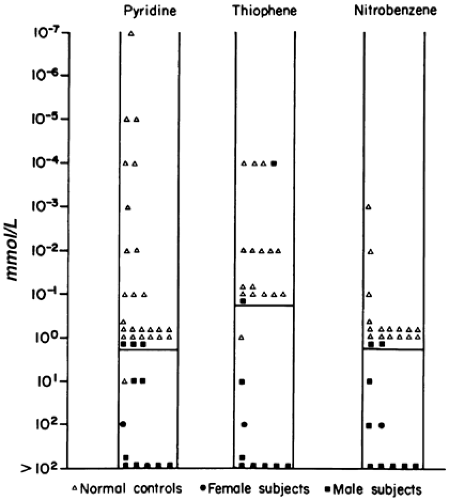 FIGURE 115-17. Olfactory thresholds for various molar concentrations of aromatic solutions in patients with hypogonadotropic hypogonadism (Kallmann syndrome). Transverse lines indicate the olfactory thresholds for normal adults. This test is performed by making separate molar solutions of the three substances indicated. The subjects are blinded to the contents of the containers and, after they are presented in random order, are asked to indicate whether a solution has an identifiable odor. Solutions should be made fresh weekly and the consistency of the investigator’s threshold should be noted to determine loss of aroma of the test solutions. The molar concentration at which the test subject can no longer consistently detect an aroma is noted. This threshold then is compared with that of a group of normal controls and to the threshold of the normal investigator. Patients with anosmia consistently fall below the normal threshold, but not all patients with hypogonadotropic hypogonadism have anosmia. (From Santen RJ, Paulsen CA. Hypogonadotropic eunuchoidism. I. Clinical study of the mode of inheritance. J Clin Endocrinol Metab 1973; 36:47.) |
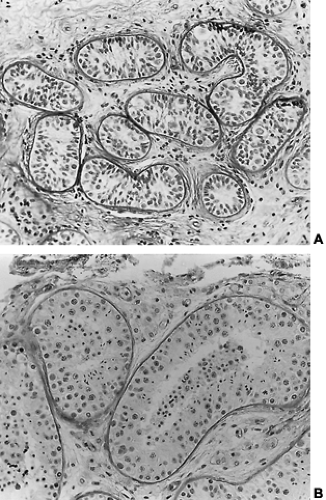 FIGURE 115-18. Initiation of spermatogenesis with administration of human chorionic gonadotropin (hCG) and human menopausal gonadotropin (hMG) in a patient with hypogonadotropic hypogonadism as demonstrated by serial testicular biopsy specimens. A, Initial state. B, After 6 months of therapy with hCG plus hMG. (From Paulsen CA. The testis. In: Williams RH, ed. Textbook of endocrinology. Philadelphia: WB Saunders, 1981:333.) |
Basal serum gonadotropin levels are low normal or undetectable in these men. When multiple samples are taken over a 24-hour period, however, some subjects demonstrate occasional small pulses of LH.144 When a single bolus dose of gonadotropin-releasing hormone is administered, the response of the pituitary gonadotropins usually is minimal. However, if repeated pulses of gonadotropin-releasing hormone are given, a normal rise in serum LH and FSH levels eventually occurs.143,145,146
These studies indicate that the most likely defect is a deficiency in LHRH secretion by the hypothalamus. Furthermore, autopsy studies have demonstrated anatomically normal pituitary glands.147 Serum testosterone levels remain in the middle of the female range in most patients with this condition before treatment, although they may have shown partial progression through puberty. No other pituitary hormonal defect has been documented in these patients.
These studies indicate that the most likely defect is a deficiency in LHRH secretion by the hypothalamus. Furthermore, autopsy studies have demonstrated anatomically normal pituitary glands.147 Serum testosterone levels remain in the middle of the female range in most patients with this condition before treatment, although they may have shown partial progression through puberty. No other pituitary hormonal defect has been documented in these patients.
Approximately 10% of men with idiopathic hypogonadotropic hypogonadism provide a history of some pubertal development with subsequent regression. This diagnosis should be suspected in any man who has passed the normal pubertal age and remains prepubertal.144 Serum gonadotropin levels are low, testosterone levels are low, and prolactin levels are normal.148,149 If one of the associated abnormalities, such as anosmia, cleft palate, and cleft lip, is present, the diagnosis can be made with some assurance. If these findings are absent, however, differentiating between delayed puberty and idiopathic hypogonadotropic eunuchoidism can be extremely difficult, because puberty does not occur until 18 or 19 years of age in some normal men. Several maneuvers have been described that purport to differentiate this condition from delayed puberty.147,150,151 These include a subnormal serum prolactin response to thyrotropin-releasing hormone or to a phenothiazine, a decreased serum testosterone response to exogenous hCG, and a normal LH response to pulsatile stimulation by LHRH. None of these tests has been consistently reliable. If the differentiation cannot be made and the circumstances warrant, treatment with testosterone or hCG for 3 or 6 months may be indicated. After this, therapy can be discontinued and the patient observed to see whether spontaneous puberty progresses.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


