Sarcomas
Charles A. Forscher
Dennis A. Casciato
I. EPIDEMIOLOGY AND ETIOLOGY.
Primary mesenchymal tumors localized outside the skeleton, parenchymatous organs, or hollow viscera are generally designated as soft tissue sarcomas (STSs). Sarcomas of the mediastinum, heart, and blood vessels are discussed in Chapter 19.
A. Incidence. Sarcomas constitute about 1% of all cancers and will account for an estimated 11,000 new cases of STS and 2,800 cases of bone sarcoma in the United States in 2011. These will be associated with 3,900 and 1,500 deaths, respectively.
1. STSs outnumber bone sarcomas by a ratio of 3:1. In children, most STSs are rhabdomyosarcomas or undifferentiated tumors originating in the head and neck regions. In adults, STSs occur most frequently on the extremities or retroperitoneum and least frequently in the head and neck region.
2. Bone sarcomas occur mostly between 10 and 20 years of age (osteogenic sarcoma) or between 40 and 60 years of age (chondrosarcoma).
3. Most sarcomas show no sexual predilection. Incidence peaks during childhood and in the fifth decade.
B. Etiology. Certain kinds of sarcomas are associated with exposure to specific agents or with underlying medical conditions:
1. Lymphangiosarcoma. Prolonged postmastectomy arm edema (Stewart-Treves syndrome)
2. Angiosarcoma and other STSs. Polyvinyl chloride, thorium dioxide, dioxin, arsenic, and androgens
3. Osteosarcoma. Radium (watch dials) exposure, postmastectomy irradiation, Paget disease of bone
4. Fibrosarcoma. Postirradiation: Paget disease of bone
5. Kaposi sarcoma. Cytomegalovirus and human immunodeficiency virus type 1 (HIV-1; discussed in Chapter 36, Section IV)
6. Leiomyosarcoma. HIV-1 in children
7. Genetic diseases and syndromes
a. Li-Fraumeni syndrome. Various sarcomas (especially rhabdomyosarcoma) and carcinomas of breast, lung, and adrenal cortex (p53 gene)
b. Neurofibromatosis. Schwannomas (NF1 gene)
c. Familial retinoblastoma. Osteosarcoma (RB1 gene)
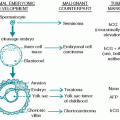
8. Chromosomal aberrations are found in nearly all sarcomas. Some of these may be limited aberrations such as specific translocations, (e.g., the X;18 translocation in synovial sarcoma, the 11;22 translocation in Ewing sarcoma, and the 12;16 translocation in myxoid liposarcoma). Other sarcomas have complex chromosomal abnormalities as occurs in myxofibrosarcoma and pleomorphic liposarcoma.
II. PATHOLOGY AND NATURAL HISTORY
A. Histology and nomenclature. Sarcomas are given a bewildering variety of names that do not indicate biologic behavior and usually do not influence therapeutic approach. The multipotential capacities of the mesenchymal tissue and
the appearance of several histologic elements in the same tumor often make clear-cut histologic diagnosis difficult.
the appearance of several histologic elements in the same tumor often make clear-cut histologic diagnosis difficult.
1. Sarcomas are named for the tissue of origin (e.g., osteosarcoma, chondrosarcoma, schwannoma, liposarcoma).
2. Tumors are also named for special histologic characteristics or given a nondescriptive name because the tissue of origin is unknown (alveolar soft parts tumor, Kaposi sarcoma, Ewing sarcoma).
3. Pathologists recognize several features in determining the grade of a sarcoma. These include the degree of cellular differentiation, the presence (or absence) of mitotic activity, spontaneous necrosis, and vascular invasion. The other descriptive terms for the tumor are far less important. Expert pathologic evaluation is crucial.
4. The presence of osteoid formation by the tumor cells suggests the diagnosis of osteogenic sarcoma. This must be distinguished from reactive or metaplastic bone formation by the pathologist.
5. Immunohistochemistry may be helpful in confirming the diagnosis of rhabdomyosarcoma and leiomyosarcoma. Expected immunophenotypes for the various sarcomas are shown in Appendix C4.III and C4.VIII.
6. Cytogenetics can be useful in the diagnosis of Ewing sarcoma, synovial sarcoma, and rhabdomyosarcoma. Newer techniques, such as fluorescent in situ hybridization (FISH) analysis, are also becoming increasingly useful.
B. Natural history. Generally, sarcomas arise de novo and not from pre-existing benign neoplasms. Tumors can “dedifferentiate,” however, from a lower grade to a higher grade. Sarcomas spread without interruption along tissue planes; they invade local nerve fibers, muscle bundles, and blood vessels. Histologic sections usually show much greater local extension than is apparent on gross examination.
1. Histologic grade. The biologic behavior of sarcomas can usually be predicted by their histologic grade. Low-grade tumors tend to remain localized; high-grade tumors (especially those with a marked degree of necrosis) have a greater propensity to metastasize. Most osteogenic sarcomas, rhabdomyosarcomas, Ewing sarcomas, and synovial sarcomas are high-grade malignancies.
2. Site of origin. The site of origin of the sarcoma may suggest the cell type as follows:
a. Head and neck
(1) Rhabdomyosarcoma (in a child)
(2) Angiosarcoma (in an elderly person)
(3) Osteogenic sarcoma (jaw)
b. Distal extremity
(1) Epithelioid sarcoma
(2) Synovial sarcoma
(3) Clear cell sarcoma
(4) Osteogenic sarcoma (femur)
c. Proximal tibia or humerus. Osteogenic sarcoma
d. Mesothelium. Mesothelioma
e. Abdomen, retroperitoneum, and mesentery
(1) Leiomyosarcoma
(2) Gastrointestinal stromal tumor (GIST; gastrointestinal sarcoma)
(3) Liposarcoma
(4) Desmoplastic small round cell tumor
f. Genitourinary tract
(1) Rhabdomyosarcoma (in a child)
(2) Leiomyosarcoma (in an adult)
g. Skin
(1) Angiosarcoma, lymphangiosarcoma
(2) Kaposi sarcoma
(3) Epithelioid sarcoma
(4) Dermatofibrosarcoma protuberans (on trunk)
3. Metastases. Sarcomas typically spread hematogenously. Lung metastases occur most commonly. Hepatic metastases can be seen from primary gastrointestinal or gynecologic sarcomas. The retroperitoneum can be a site of metastasis for extremity liposarcomas. Other sites, such as bone, subcutaneous tissue, and brain, are less common and are often detected only after pulmonary metastases have developed (tertiary metastases).
a. Sarcomas that metastasize to lymph nodes
(1) Rhabdomyosarcoma
(2) Synovial sarcoma
(3) Epithelioid sarcoma
b. Sarcomas that rarely metastasize and are associated with a favorable survival
(1) Liposarcoma (well-differentiated types)
(2) Fibrosarcoma (infantile and well-differentiated types)
(3) Malignant fibrous histiocytoma/myxofibrosarcoma (superficial type)
(4) Dermatofibrosarcoma protuberans
(5) Parosteal osteosarcoma
(6) Kaposi sarcoma when not related to acquired immunodeficiency syndrome (AIDS)
4. Paraneoplastic syndromes associated with sarcomas
a. Hypoglycemia (particularly with retroperitoneal fibrosarcoma)
b. Hypertrophic osteoarthropathy (sarcoma of pleura or mediastinum)
c. Hypocalcemia
d. Oncogenic osteomalacia
C. Clinical aspects of specific STSs
1. Alveolar soft-part sarcoma
a. Tissue of origin (incidence). Unknown (rare)
b. Features. Unique histology with no benign counterpart; often indolent even with lung metastases, which are common. The sarcoma most associated with brain metastases. Commonly affects the thigh in adults and the head and neck in children. The 5-year survival rate exceeds 60%.
2. Angiosarcoma (hemangiosarcoma, lymphangiosarcoma)
a. Tissue of origin (incidence). Blood or lymph vessels (2% to 3%)
b. Features of hemangiosarcoma. Affects the elderly; aggressive. Arises in many organs, notably the head and neck region, breast, and liver; especially affects the skin and superficial soft tissues (whereas most STSs are deep). Dedifferentiation from a hemangioma is rare. The 5-year survival rate is <20%.
c. Features of lymphangiosarcoma. Affects older adults; aggressive. Arises in areas with chronic lymphatic stasis (especially postmastectomy). The 5-year survival rate is 10%.
3. Clear cell sarcoma
a. Tissue of origin (incidence). Now recognized as a form of malignant melanoma (rare)
b. Features. Affects adults <40 years of age; painless, firm, spherical masses on tendon sheaths and aponeurotic structures of distal extremities. The 5-year survival rate is about 50%.
4. Epithelioid sarcoma
a. Tissue of origin (incidence). Unknown (rare)
b. Features. Affects young adults; aggressive; typically appears on distal extremities. Epithelioid sarcoma and synovial sarcoma are the most common tumors of the hand and foot. Differs from other STSs by having a greater tendency to spread to noncontiguous areas of skin, subcutaneous tissue, fat, draining lymph nodes, and bone. The 5-year survival rate is about 30%.
5. Fibrosarcoma
a. Tissue of origin (incidence). Fibrous tissue (5% to 20%)
b. Features. Affects all age groups; arises in many mesenchymal sites; usually involves the abdominal wall or extremities. Ninety percent are well differentiated (desmoid). Dermatofibrosarcoma protuberans (rare) develops on the skin of the trunk and almost never metastasizes. Fibromyxosarcoma affects any soft tissue site but usually develops on the extremities. Ten percent are poorly differentiated (high grade). Survival is directly related to tumor grade (also see Section II.D.5).
6. Malignant fibrous histiocytoma (MFH)/myxofibrosarcoma. Many pathologists now prefer the term myxofibrosarcoma for these tumors. Some tumors previously felt to represent MFH are now classified as pleomorphic liposarcomas or pleomorphic sarcoma, not otherwise specified (NOS).
a. Tissue of origin (incidence). Unknown (10% to 23%)
b. Features. Age > 40 years (<5% of affected patients are <20 years of age). MFH has become a common histologic diagnosis. Develops in extremities (especially legs), trunk, and retroperitoneum. Superficial MFH develops close to the skin surface and is often low grade; the 5-year survival rate is 65%. Deep MFH usually is high grade; the 5-year survival rate is 30% to 60%.
7. Hemangiopericytoma/solitary fibrous tumor
a. Tissue of origin (incidence). Blood vessels or fibrous tissue (<1%)
b. Features. Affects all ages. Develops under finger tips (glomus tumors), on lower extremities or pelvis, in the retroperitoneum, and elsewhere. Benign and malignant versions. The 5-year survival rate is about 50%.
8. Kaposi sarcoma (KS). All varieties of KS are associated with human herpesvirus type 8 (HHV-8). KS typically presents as purplish blotches or nodules that may be painful or pruritic. Treatment of KS in patients with AIDS is discussed in Chapter 36, Section IV.
a. Tissue of origin (incidence). Controversial (varied)
b. Features of classic KS. Classically affects older adults with Mediterranean ancestry; extremely indolent lesions arise on lower extremities (occasionally on hands, ears, and nose) and rarely cause death.
c. Features of epidemic KS. The epidemic and aggressive variety is associated with AIDS (see Chapter 36, Section IV), African children, renal transplant recipients, immunosuppressed nontransplantation patients, and Eskimos. These patients develop a widely disseminated, aggressive, and usually fatal form of the disease. Generalized cutaneous involvement, generalized lymphadenopathy, and visceral or gastrointestinal involvement are typical.
9. Leiomyosarcoma, gastrointestinal stromal tumor (GIST), and metastasizing leiomyoma
a. Tissue of origin (incidence). Smooth muscle for leiomyoma and leiomyosarcoma; interstitial cell of Cajal for GIST (7% to 11%)
b. Features. Affects all age groups. Develops in gastrointestinal tract, uterus, retroperitoneum, and other soft tissues. Generally refractory to chemotherapy and radiotherapy. The 5-year survival rate is 30%.
c. Gastrointestinal stromal tumors (GISTs) are morphologically similar to leiomyosarcoma but have different immunohistochemical staining characteristics (see Appendix C4.III). GISTs do not stain for actin (as leiomyosarcomas do), and most express CD117 (c-kit protein). Treatment of GIST is presented in Section VII.C.7.
d. Leiomyoma peritonealis disseminata (LPD). A condition in women, usually in reproductive years. Myriads of asymptomatic benign leiomyomas are usually scattered throughout the peritoneal cavity and range from 1 to 10 cm in size; they are stimulated by estrogen. LPD causes occasional mechanical problems with bowel or pain. Generally, no treatment is required; when symptomatic, treatment is with estrogens or antiestrogens.
e. Leiomyoma, benign metastasizing. Histologically benign, these leiomyomas are typically discovered as persistent pulmonary nodules and possibly as a variant of LPD. Associated nodules in the pelvis are mostly in round ligaments of uterus and not as diffuse as in LPD. Treatment is surgical for symptomatic or progressive lesions.
10. Liposarcoma
a. Tissue of origin (incidence). Fat tissue (15% to 18%)
b. Features. Affects middle and older age groups, mostly men. Develops in the thigh, groin, buttocks, shoulder girdle, and retroperitoneum. Does not arise from benign lipomas. Low-grade tumors in the extremity are now called atypical lipomatous tumor. The designation of well- differentiated liposarcoma remains for tumors of the abdomen and retroperitoneum. The 5-year survival rate is 80% for low-grade liposarcomas and 20% for high-grade liposarcomas of an extremity. Survival rates are lower and local recurrence rates are high for abdominal/retroperitoneal liposarcomas.
11. Mesothelioma
a. Tissue of origin (incidence). Mesothelium
b. Features. Age > 50 years. Asbestos exposure is etiologic. Involves pleura and peritoneum; aggressively encases viscera. Highly lethal; the 5-year survival rate is <10% (see Chapter 8).
12. Myxoma
a. Tissue of origin (incidence). Mesenchymal tissues
b. Features. Usually found on extremities; has histologic appearance of umbilical cord. The 5-year survival rate is about 80%.
13. Neurofibrosarcoma (schwannoma, neurilemoma)
a. Tissue of origin (incidence). Nerve (5% to 7%)
b. Features. Affects young and middle-aged adults and patients with neurofibromatosis type 1 (von Recklinghausen disease; about 10% develop sarcomatous changes during lifetime). Histologically resembles fibrosarcoma. Presents with thickening of nerves and without anatomic predilection. Superficial variety is low grade, spreads extensively along nerve sheaths without metastasizing, and has a 5-year survival rate of >90%. Penetrating variety is high grade with nodular growth, vascular invasion, and lung metastases and has a 5-year survival rate of <20%.
14. Rhabdomyosarcoma
a. Tissue of origin (incidence). Striated muscle (5% to 19%)
b. Features. By definition in the G-TNM staging system, all are grade 3. All types can occur in any age group, but the typical onset and
distribution is noted below (see Chapter 18, Cancers in Childhood, “Rhabdomyosarcoma”).
distribution is noted below (see Chapter 18, Cancers in Childhood, “Rhabdomyosarcoma”).
c. Features of embryonal rhabdomyosarcoma. Affects infants and children; sites are head and neck (70%) and genitalia (15% to 20%). Includes sarcoma botryoid. The 5-year survival rate is about 70%.
d. Features of alveolar rhabdomyosarcoma. Affects teenagers at any site; highly aggressive; histology resembles lung alveoli. The 5-year survival rate is about 50%.
e. Features of pleomorphic rhabdomyosarcoma. Affects patients >30 years of age, is rare, and develops in extremities. Often is highly anaplastic; microscopically confused with MFH. The 5-year survival rate is about 25%.
15. Synovial sarcoma
a. Tissue of origin (incidence). Unknown. Recent data suggests these tumors may arise from primitive muscle cells. Although these tumors can arise near joints, they are not composed of cells with synovial differentiation. The name is a misnomer that persists. They rarely arise within a joint space.
b. Features. Affects young adults but may occur from the second to fourth decade. Monophasic and biphasic subtypes are distinguished. Presents with hard masses, often painful, near tendons in the vicinity of joints of the hands, knees, or feet. Synovial and epithelioid sarcomas are the most common tumors of the hand and foot. Often calcified, with characteristic radiographic appearance. The majority of synovial sarcomas are high grade. Lymph node involvement may be seen in up to 20% of cases. The 5-year survival rate is from 30% to 50%.
D. Clinical aspects of specific bone sarcomas
1. Adamantinoma
a. Tissue of origin (incidence). Unknown; nonosseous (<1%)
b. Features. Osteolytic tumor; often develops on upper tibia; resembles ameloblastoma of mandible. Indolent behavior; the 5-year survival rate is >90%.
2. Chondrosarcoma
a. Tissue of origin (incidence). Cartilage (30%)
b. Features. Age 40 to 60 years; <4% of patients are <20 years of age. Usually develops in shoulder girdle (15%), proximal femur (20%), or pelvis (30%). Chondrosarcomas are the most common malignant tumors of the sternum and scapula. Most tumors are grade 1 or 2; higher grade tumors metastasize frequently; however, tumor grade does not appear to affect prognosis. Local recurrence is a major problem in management. Usually refractory to both radiation therapy (RT) and chemotherapy. Dedifferentiated chondrosarcomas may, however, respond to chemotherapy. Complete surgical removal is the main determinant of recurrence and survival. The 5-year survival rate is about 50%.
(1) Central chondrosarcomas (75%) arise within a bone; peripheral chondrosarcomas (25%) arise from the surface of a bone. Peripheral lesions can become quite large without causing pain; central lesions present with a dull pain but rarely with a mass. Pain means that the apparently “benign” cartilage tumor on radiographs is probably a central chondrosarcoma.