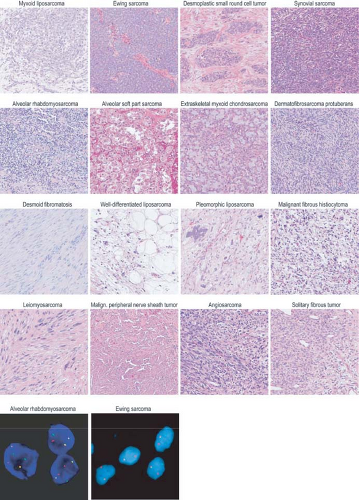
 FIGURE 26.1 Sarcoma subtypes discussed in the text. Upper panels, hematoxylin and eosin-stained paraffin sections. Malign., malignant. Lower panels are fluorescence in situ hybridization images showing (left) alveolar rhabdomyosarcoma with fusion of probes for PAX3 (red) and FOXO1 (green) and (right) Ewing sarcoma with break-apart of probes flanking the EWS breakpoint region, EWSR1. |
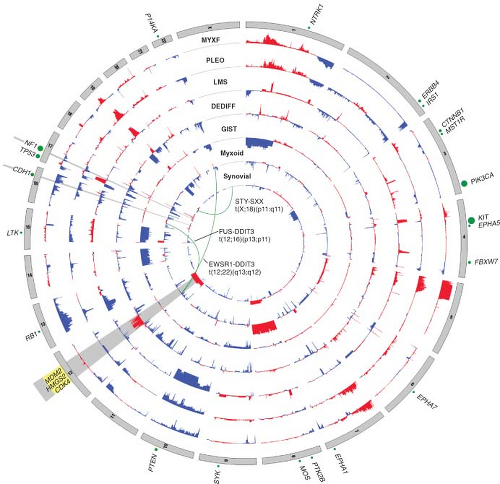 FIGURE 26.2 Nucleotide and copy number alterations in soft tissue sarcoma. The outer ring indicates chromosomal position. The second through fifth rings represent four subtypes with complex karyotypes (as labeled; MYXF, myxofibrosarcoma; PLEO, pleomorphic liposarcoma; LMS, leiomyosarcoma; DEDIFF, dedifferentiated liposarcoma). The three inner rings represent subtypes with simple karyotypes (Myxoid, myxoid/round cell liposarcoma). The plots show the statistical significance of genomic aberrations, with amplification in red and deletion in blue. Green curves indicate the chromosomal breakpoints of pathognomonic translocations in myxoid/round-cell liposarcoma and synovial sarcoma. Genes harboring somatic nucleotide alterations are indicated with green circles whose size is proportional to their frequency of occurrence. (Courtesy of Barry S. Taylor, Computational Biology Center, Memorial Sloan-Kettering Cancer. Adapted from ref. 19.) |
TABLE 26.1 CYTOGENETIC AND MOLECULAR ABNORMALITIES IN SOFT TISSUE SARCOMAS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
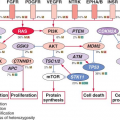
be either bone or soft tissues. A range of aggressive small, blue, round cell tumors with variations in clinical and morphologic features have been subsumed under the general term Ewing sarcoma family tumor following the recognition of common pathognomonic chromosomal translocations.23,24 EWSR1, the common 5′ translocation partner, is fused to one of several possible ETS family transcription factor genes (usually FLI1). In the fusion protein, EWSR1 provides, at minimum, its 264 amino acid N-terminal transcriptional regulatory domain, and the ETS factor provides its C-terminal DNA-binding domain. In the process, EWSR1 loses its RNA recognition domain, and the ETS factor loses its native transactivation domain. Several direct transcriptional targets for the fusion oncoprotein are supported by strong evidence. Some of these targets are up-regulated (ID2,25 PTPL1,26 MK-STYX,27 DAX128) and some repressed (CIP1,29 TGFBR2,30 IGFBP331) in Ewing sarcoma, but gene repression events, mediated by cofactors, appear to predominate overall.32 The net result is activation of pathways driving proliferation and cell survival (including IGF signaling33), with concurrent repression of pathways promoting mesenchymal differentiation.34,35
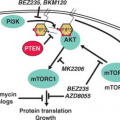
potential value for IGF/Akt/mTOR inhibitors in this disease.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


